The human body contains at least 20,000 different proteins, often called the “workhorses of the cell” because of their role in keeping cells healthy. Each protein consists of a unique string of amino acids that affects its shape and function—or dysfunction, in the case of proteins that assemble incorrectly, which can cause disease.
By understanding and predicting the vast array of shapes a protein can take, scientists can design drugs that target specific proteins with specific roles in a cell. The hope is that technologies like Google’s AlphaFold—which uses artificial intelligence (AI) to predict the structure of proteins, DNA and other biomolecules—will speed up this daunting task and subsequently the development of potentially lifesaving medications.
University of Maryland researchers are “cautiously optimistic” about this ambitious goal but say that AlphaFold must be paired with a stronger foundation of physics to be successful. A method they developed, described in a new paper published in the journal eLife, does just that.
“There are lots of uncured diseases, and we hope that AI can help us screen a large number of compounds to identify effective, non-toxic drugs in a cost-efficient manner, ultimately lowering health care costs for all,” said the study’s senior author, Pratyush Tiwary. “Our method will speed up drug discovery and enable personalized medicine for complex diseases.”
Tiwary is a professor in the University of Maryland, College Park’s Department of Chemistry and Biochemistry and Institute for Physical Science and Technology (IPST) and the University of Maryland Institute for Health Computing (UM-IHC).
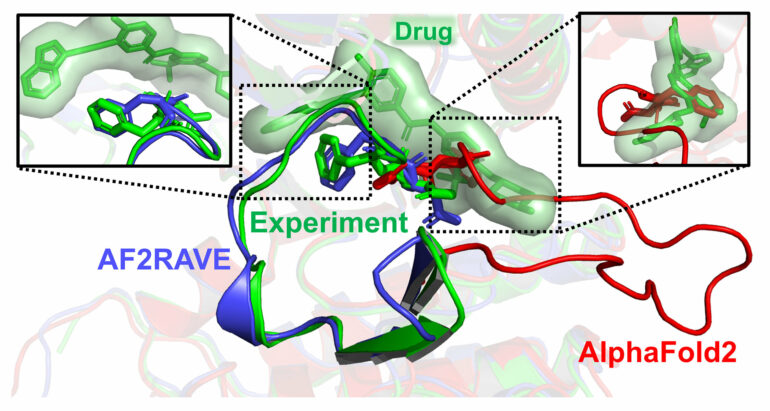
Their method, AlphaFold2-RAVE (AF2RAVE), fuses AlphaFold’s strengths with computer simulations that are based on the laws of physics. It expands on an earlier technique called RAVE, designed by Tiwary and his students in 2018 to speed up molecular simulations that typically take a long time to process.
AlphaFold excels at predicting the structure of proteins in their “native”—or folded—state, but it cannot predict proteins in a non-native state. Much like origami, proteins can fold into different shapes, with those folds affecting a protein’s ability to carry out a task.
In a non-native form, proteins can be unfolded or misfolded, which limits their function and can even lead to diseases like Alzheimer’s and Parkinson’s. These non-native shapes are less predictable but still crucial to the development of new pharmaceuticals.
Tiwary explained that AlphaFold overlooks non-native structures because it relies on information deposited in the Protein Data Bank, a database that primarily contains the structures of proteins in their native state.
“What AlphaFold did—and this was a breakthrough—was predict the most stable structure a protein can take,” said Tiwary, who also holds the Millard and Lee Alexander Professorship in Chemical Physics at UMD. “What it cannot do is predict the other structures a protein can take. This is a fundamental problem with AI—not just in chemistry, but across all disciplines— because AI methods are only as good as their training database.”
To overcome this problem, Tiwary and his co-authors applied their RAVE method to AlphaFold 2, which was the most recent version of the technology before Google released AlphaFold 3 in May 2024.
“Our method begins with generating thousands of possible hypotheses, or protein structures, using AlphaFold 2, analogous to repeatedly querying an AI tool like ChatGPT for answers,” Tiwary explained. “These hypothetical structures are then evaluated through high-resolution computer simulations based on the laws of physics, particularly thermodynamics and statistical mechanics.”
They used their method to generate possible native and non-native structures of three kinases, a type of protein that plays an important role in cell growth. Abnormal kinase activity can lead to the development of cancers, which partly explains why kinases are the second most important drug target—second only to G protein-coupled receptors, a family of proteins with diverse functions.
In a first, the researchers were able to predict the 3D structures that kinases could take and rank them thermodynamically, ultimately revealing the top two non-native structures out of 1,000 that drug candidates could potentially bind to. Thermodynamics is relevant to drug design because it dictates the likelihood that a particular protein shape will appear in nature, as well as how successfully a drug might bind to a protein, increasing its effectiveness.
“This evaluation determines the probability of each structure forming under room temperature and pressure,” Tiwary said. “As a result, the thousands of initial hypotheses are filtered down to a more manageable and likely set of structures.”
Their method could reduce the need for drug docking, a computational method that is used to determine how a drug molecule might bind to a particular protein. When the researchers compared AF2RAVE’s list of drug candidates with existing drugs that are known to target a particular kinase shape, the researchers learned that their results matched more than 50% of the time—a success rate that could not have been achieved without this method, Tiwary said.
“The success rate is almost nonexistent when using AlphaFold alone,” Tiwary said. “Without our approach, you would have to dock drugs to each of the 1,000 structures generated by AlphaFold 2. For some kinases, 1 out of 1,000 is a good drug candidate, but for others, none of the 1,000 are suitable.”
Although the researchers only applied their method to AlphaFold 2, Tiwary explained that the newest version of AlphaFold does not resolve the previous issues with non-native protein structures.
By coupling AI with physics, the AF2RAVE method could usher in a new era of personalized therapy by identifying drugs that are tailored to patients’ unique genetic profiles. It could also lead to new drugs that bind to non-native proteins, which are trickier to develop.
This discovery is just the beginning of what Tiwary and his colleagues will be able to accomplish with their AF2RAVE method.
“This study—and what we are going to do next—represents the best of UM-IHC and IPST,” said Tiwary, who leads the therapeutic target discovery research center at UM-IHC. “It’s rigorous scientific computing with practical implications, and all of it’s grounded in statistical mechanics, which IPST has been a leader in for the last several decades. This intersection is an incredibly exciting place to be!”

Tiwary is working with UM-IHC Co-Executive Director Bradley Maron and David Weber at the Institute for Bioscience and Biotechnology Research to apply AF2RAVE to hypertension and Alzheimer’s disease in hopes of identifying better treatments.
“By using AI to predict multiple protein configurations, this revolutionary AF2RAVE method has the potential to speed up drug innovation by identifying novel targets that determine the effect of protein function on cellular mechanisms underpinning disease,” said Maron, who is also a professor of medicine at the University of Maryland School of Medicine. “We are on the verge of a new era that will usher in treatments targeting these complex protein folding processes.”
In addition, Tiwary and his team are working with National Cancer Institute researchers Gregoire Altan-Bonnet, Naomi Taylor and John Schneekloth to apply these methods to pediatric immunotherapy and cancer.
In these projects and future ones, Tiwary believes that AI can ethically be applied to the fields of biophysics and biochemistry to better people’s lives.
“AI, especially when guided by physics, is here to stay,” Tiwary said. “AF2RAVE is not just another method—this is something that is going to have an impact on people’s day-to-day lives.”
More information:
Xinyu Gu et al, Empowering AlphaFold2 for protein conformation selective drug discovery with AlphaFold2-RAVE, eLife (2024). DOI: 10.7554/eLife.99702.1
Provided by
University of Maryland
Citation:
Scientists ‘cautiously optimistic’ about AI’s role in drug discovery (2024, August 2)

