The completion of the full “telomere-to-telomere” (T2T) human genome last year emphasized that genome sequences that were previously thought to be “complete” were not, in fact, complete at all.
Moreover, many recent genomes are sequenced with short-read sequencing technologies, which fragment DNA into short segments, typically 150-300 base pairs long, and are then compared to a reference sequence. While fast, accurate and relatively economical, short-read methodologies routinely miss large parts of the genome, about 10% overall. The missing segments include regions of high G/C content and repetitive sequences, including segmental duplications, simple repeats, and transposable elements (TEs).
TEs are repetitive sequences that have moved to other locations in the genome, and the mobility of these sequences contribute greatly to genomic variation. Repetitive sequences frequently underlie the formation of structural variants (SVs)- genomic differences resulting from duplications, insertions, deletions, and inversions. SVs are often missed when using short read sequencing (in particular those mediated by repeats) but they can play important roles in genome dysregulation and disease.
Researchers have turned to long-read sequencing to more completely analyze genomes, as these technologies enable sequencing of far longer DNA segments and can accurately capture a more complete picture of a genome. Recent advances have improved long read accuracy and utility, allowing researchers to investigate previously undetected genomic features, and not just in humans.
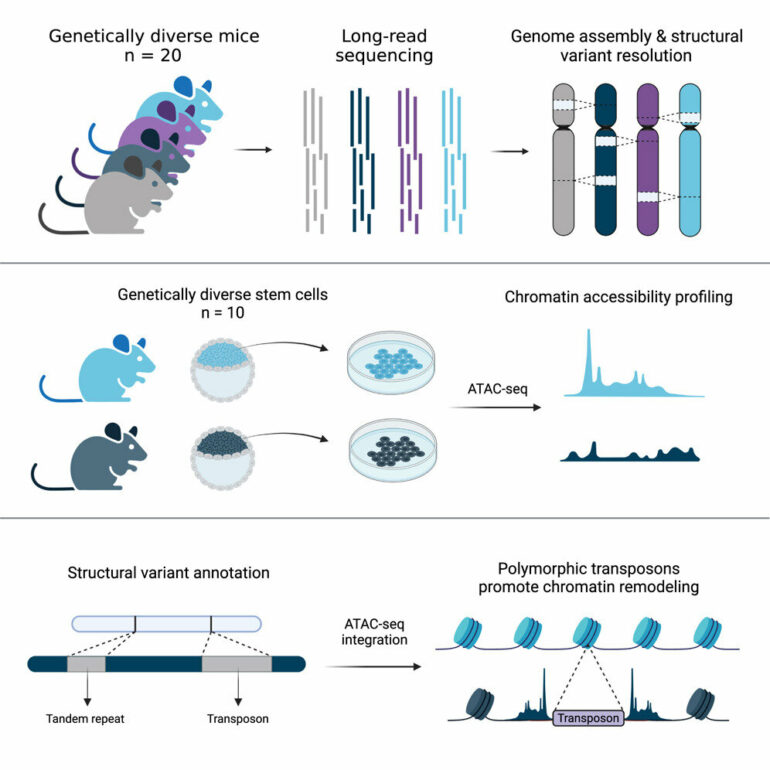
Jackson Laboratory (JAX) and University of Connecticut Health Center Assistant Professor Christine Beck, Ph.D., led a team that explored the genomes of another notable species, the mouse, and revealed details across 20 diverse inbred strains that will be critical for informing mouse-based genetics and genomics research moving forward.
Structural variation between mouse strains
Mice have their own reference genome, known as GRCm39, based on the sequence of C57BL/6J, a strain from the Mus musculus domesticus subspecies. But many commonly used laboratory mouse strains have been derived from two other subspecies as well, Mus musculus castaneus and Mus musculus musculus, and there are many genetic differences between different inbred strains.
For the work presented in “Resolution of structural variation in diverse mouse genomes reveals chromatin remodeling due to transposable elements,” published in Cell Genomics, Dr. Beck selected a wide variety of commonly used strains, including the seven parental founders of the genetically diverse Collaborative Cross (CC) and Diversity Outbred (DO) mouse panels, six resultant CC strains with abnormalities of unknown genetic origin, and seven other commonly used strains with different genetic backgrounds.
Ardian Ferraj, a graduate student and the lead author on the study, then assembled the genomes of these 20 mice, and used these sequences to identify SVs present in the animals that differentiated their genomes from that of the C57BL/6J reference. Using PAV, a program developed by Beck lab member Dr. Peter Audano, Ardian showed that SVs are prevalent across mouse genomes and contribute extensively to genomic variation. In fact, SVs contain nearly five times the number of bases affected compared to previously published single nucleotide variants from diverse mouse genomes.
They also found a much greater diversity from SVs between mouse genomes than between human genomes, suggesting that a single mouse reference genome is inadequate for mapping genomic data across mouse strains. Importantly, long-read sequencing is vital for capturing this variation. Across 18 of the mouse strains, the research team detected an additional 213,688 insertions, 64,277 deletions and 97 inversions with long reads compared to short-read data.
. DOI: 10.1016/j.xgen.2023.100291")
© Cell Genomics (2023). DOI: 10.1016/j.xgen.2023.100291
Transposable elements and structural variation consequences
While only a small number of TEs are still able to mobilize in human genomes, they are more mobile in mice. Because of this, Beck and her team focused on transposable element variants (TEVs), which they found comprised nearly 40% of all SVs, with most (60%) being insertions. There are multiple kinds of TEVs, known as short versus long interspersed nuclear elements (SINEs and LINEs), which are predictably characterized by their size. LINEs were nearly twice as common as SINEs in the mouse genomes, 47% to 24%.
Because of their size, LINEs also contribute nearly half of variable sequence content in mouse genomes, compared to just 24% contributed by non-TEV SVs and 2.1% by SINEs. Various endogenous retroviral sequences generated the remaining 28% of TEVs. Retroviruses are RNA viruses whose genomes are reverse transcribed to DNA, which is then inserted into the genome. While many current retroviruses are associated with diseases such as AIDS and cancer, normal mammalian genomes contain large amounts of DNA derived from retroviruses over the millennia, known as endogenous retroviruses or ERVs, that help drive genomic variation in mice.
So what are the possible consequences of all this genomic variation and activity? The researchers looked at the SVs in the context of known genomic features and predicted severity of effects. Among the newly detected SVs within gene sequences, the vast majority (94,863) were within introns, the sequences that are spliced out of pre-mRNAs so they don’t alter protein structure; 1,469 were in the untranslated segments (UTRs) at either end of the gene; and 510 within the actual protein coding sequences.
They also identified a previously undetected retroviral element insertion within a specific gene, Mutyh, a DNA repair gene associated with a known mutational signature in certain mouse strains. The underlying variant was unknown, but the team found that the insertion was associated with a significant decrease in Mutyh gene expression. The finding shows that unknown SVs can alter important genomic regions and reside in genes associated with traits relevant to health and function, including disease.
Finally, in collaboration with Jax investigator Dr. Laura Reinholdt, the team investigated the impact of TEs on embryonic stem cell differences. TEs promote genome diversity and their variation may alter important aspects of gene expression between strains. Indeed, the study found more than 22,000 TEVs associated with significant changes in stem cell chromatin accessibility, a key regulator of gene expression, across embryonic stem cells from 10 genetically diverse mouse strains.

Again focusing on a specific example, they investigated a strain-specific (CAST/EiJ) intronic insertion in the gene Slc47a2, which was accompanied by a chromatin accessibility signal unique to the strain. They found elevated levels of Slc47a2 expression compared to strains lacking the insertion, with a strain-specific transcript and a possible binding region for a pluripotency factor, indicating important roles for TEVs in early development.
A more complete understanding
Given the importance of the mouse as a model for mammalian genetics and human disease, it’s necessary to fully understand the functional consequences of genomic variation. The comprehensive detection and characterization of SVs between mouse strain genomes is a crucial part of such understanding, and the results and data generated by Dr. Beck and her collaborators provide an important step forward for the field.
The authors produced a sequence-resolved SV resource, a mouse embryonic stem cell expression resource, and chromatin accessibility data for the research community that may help further investigations into mouse evolution and the genomics underlying traits of interest.
More information:
Christine R. Beck, Resolution of structural variation in diverse mouse genomes reveals chromatin remodeling due to transposable elements, Cell Genomics (2023). DOI: 10.1016/j.xgen.2023.100291. www.cell.com/cell-genomics/ful … 2666-979X(23)00057-5
Provided by
Jackson Laboratory
Citation:
New study reveals details across 20 diverse inbred mouse strains (2023, April 5)

