Since the first sequencing of the human genome more than 20 years ago, the study of human genomes has relied almost exclusively on a single reference genome to which others are compared to identify genetic variations. Scientists have long recognized that a single reference genome cannot represent human diversity and that using it introduces a pervasive bias into these studies. Now, they finally have a practical alternative.
In a paper published December 16 in Science, researchers at the UC Santa Cruz Genomics Institute have introduced a new tool, called Giraffe, that can efficiently map new genome sequences to a “pangenome” representing many diverse human genome sequences. They show that this approach allows a more comprehensive characterization of genetic variations and can improve the genomic analyses used by a wide range of researchers and clinicians.
“We’ve been working toward this for years, and now for the first time we have something practical that works fast and works better than the single reference genome,” said corresponding author Benedict Paten, associate professor of biomolecular engineering at UC Santa Cruz and associate director of the Genomics Institute. “It’s important for the future of biomedicine that genomics helps everyone equally, so we need tools that account for the diversity of human populations and are not biased.”
All humans have the same genes, but there are many variations in the exact sequences of the genes—meaning the sequence of DNA subunits (abbreviated A, C, T, G) that spell out the genetic code—as well as in the vast stretches of the genome outside of the protein-coding genes. A difference in a single letter of code is called a single nucleotide variant (SNV), and insertions or deletions of short sequences are known collectively as “indels”.
The most complex variants are structural variations involving rearrangements of large segments of code (50 or more letters). These are especially hard to find using a single reference genome, yet they can have significant effects and are known to play an important role in some diseases. The average person has millions of SNVs and indels and tens of thousands of larger structural variants, and collectively the structural variants actually involve more letters of code than the other types of variants do.
“The workhorses of genomics have been SNVs and short indels, because structural variants have been hidden from view,” Paten said. “Pangenomics is making structural variants visible so we can study them the same way we do SNVs and short indels. There are a lot of structural variants and they can have a big impact, so this is critical for the future of genetic studies of disease.”
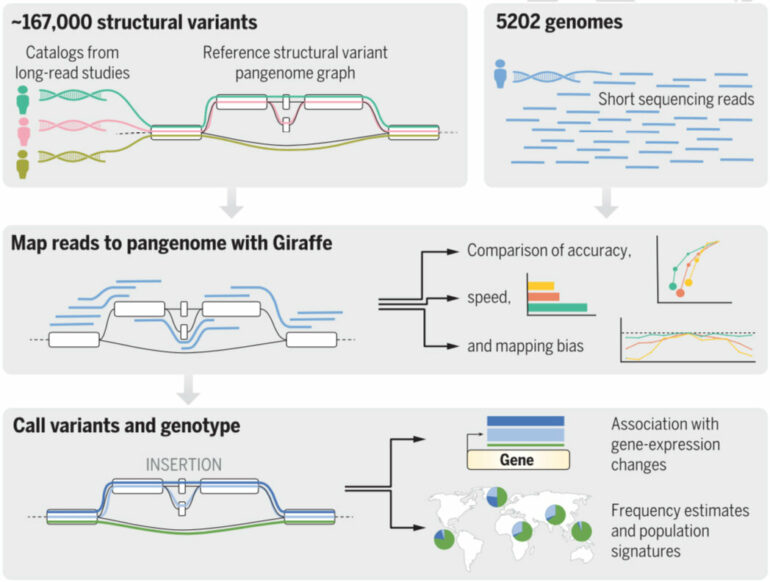
A pangenome reference can be created from multiple genome sequences using a mathematical graph structure to represent the relationships between different sequences. In the new paper, the researchers built two human genome reference graphs using publicly available data. These were used to evaluate the new tool, Giraffe, which is a set of algorithms for mapping new sequence data to a pangenome reference.
First author Jouni Sirén, a research scientist at the Genomics Institute, pioneered many of Giraffe’s key algorithmic innovations. Giraffe can accurately map new sequence data to thousands of genomes embedded in a pangenome reference as quickly as existing tools map to a single reference genome. The study also showed that using Giraffe reduces mapping bias, the tendency to incorrectly map sequences that differ from the reference genome.
. Giraffe (and competing mappers) mapped reads to the graph or to linear references, and mapping accuracy, allele coverage balance, and speed were evaluated (middle). Then, mapped reads were used for variant calling, and variant call accuracy was evaluated (bottom). Structural variant calls were analyzed alongside expression data to identify eQTLs and population frequency estimates. © Sirén et al., Science 2021")
Overview of the experiments: Variant calls from long read–based and large-scale sequencing studies were used to construct pangenome reference graphs (top). Giraffe (and competing mappers) mapped reads to the graph or to linear references, and mapping accuracy, allele coverage balance, and speed were evaluated (middle). Then, mapped reads were used for variant calling, and variant call accuracy was evaluated (bottom). Structural variant calls were analyzed alongside expression data to identify eQTLs and population frequency estimates. © Sirén et al., Science 2021
“Not only is the analysis better, it is also as fast as current methods that use a linear reference genome,” said co-first author Jean Monlong, a postdoctoral researcher at the Genomics Institute.
Inexpensive short-read sequencing is a mainstay of modern genomics, yielding snippets of sequence that must be mapped to a reference genome to make sense of them. Mapping shows where each snippet belongs on one of the 23 human chromosomes and identifies the variants present at each location in an individual’s genome, a process known as genotyping.
The researchers found that Google Health’s deep-learning variant caller, DeepVariant, could more accurately identify SNVs and indels using Giraffe’s alignments against a pangenome than it could using alignments against a single reference genome.
Monlong said he was most excited about using pangenomics to study structural variants.
“A lot of structural variants have been discovered recently using long-read sequencing,” he said. “With pangenomes, we can look for these structural variants in large datasets of short-read sequencing. It’s exciting because this will allow us to study those new structural variants across many people and ask questions about their functional impact, association with disease, or role in evolution.”
The researchers used Giraffe to map sequence reads from a diverse group of 5,202 people and determine their genotypes for 167,000 recently discovered structural variations. This enabled them to estimate the frequency of different versions of these structural variants in the human population as a whole and within individual subpopulations. They showed that the frequency of some variants differs considerably between subpopulations and could be misinterpreted if analyzed only in, for example, European-ancestry populations where the frequency of a particular variant is low.

A single reference genome must choose one version of any variation to represent, leaving the other versions unrepresented. By making more broadly representative pangenome references practical, Giraffe can make genomics more inclusive.
Paten and others at the UC Santa Cruz Genomics Institute are involved in a major effort funded by the National Human Genome Research Institute to build a comprehensive human pangenome reference, which they expect to release next year as a resource for the scientific community.
In addition to Sirén and Monlong, the new paper has three other co-first authors who contributed equally: Xian Chang, Adam Novak, and Jordan Eizenga, all at the UC Santa Cruz Genomics Institute. In addition to other coauthors at the Genomics Institute, including Director David Haussler, the coauthors also include researchers at Google Health, Broad Institute of MIT and Harvard, University of Michigan, University of Virginia, Harbor-UCLA Medical Center, and University of Tennessee Health Science Center.
More information:
Jouni Sirén et al, Pangenomics enables genotyping known structural variants in 5,202 diverse genomes, Science (2021). DOI: 10.1126/science.abg8871. www.science.org/doi/10.1126/science.abg8871
Provided by
University of California – Santa Cruz
Citation:
A new way to find genetic variations removes bias from human genotyping (2021, December 16)

