DNA sequencing has changed biology like nothing else since the origin of species theory. In particular, the way we investigate microbial life has fundamentally changed. Today, we are able to sequence DNA with unprecedented speed and resolution, so that we are even able to sequence genomes of microbes that have never been described or cultivated before. At the same time, whole-genome sequencing of known—most pathogenic—species, has become a routine methodology carried out worldwide as a daily business.
This, in turn, constantly increases the amount of publicly stored sequences, which are equally becoming a treasure trove and a hurdle both at the same time. For many sequence-based computational analyses, comprehensive and thorough genome annotations play a crucial role as a common starting ground. And for a long time this has been perceived as a solved problem.
But, the daily influx of new genome and gene sequences into public databases poses new issues for the rapid annotation of microbial genomes. In particular, the search for similar or identical protein-coding genes has become a large-scale bioinformatics search problem like a needle in a haystack—an astonishingly large haystack, nowadays.
In this context, we’re facing two diametrically diverging developments. On one hand, public databases are flooded with similar and near-identical protein sequences. For instance, these include those of utmost relevance like antimicrobial resistance genes and virulence factors—sequences which can be crosslinked with tons of useful information from many public databases. On the other hand, countless new sequences emerge from metagenome projects sequencing of what is often referred to as microbial dark matter. However, for many of these sequences no additional information is available at all.
Two distinct bioinformatic challenges arise from this situation: first, the exact identification of known sequences, and second, the functional description of rare or even unknown sequences—both in the order of hundreds of millions. To address these challenges, we tried an alignment-free protein sequence hashing strategy coupled with two hierarchical sequence alignment steps as a new approach to this problem. Our work was published in the journal Microbial Genomics.
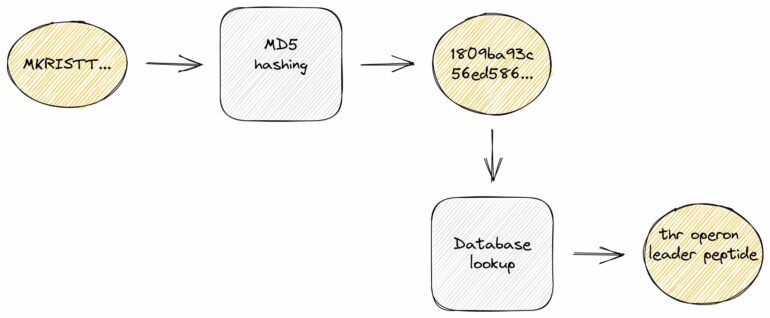
To exactly identify known protein sequences, we used a hash function that maps input data of arbitrary lengths to fixed-size binary fingerprints. These hash functions are well-known from so-called checksum calculations due to an important characteristic: they are extremely fast to compute, much faster than traditional sequence alignments.
To take advantage of this, we created a compact, local database with hash fingerprints of more than 220 million protein sequences. In a second step, we pre-assigned high-quality annotations and cross-links to further external databases. Of note, these demanding large-scale computations are only required once at the database compilation step which we regularly conduct upon new releases. For the actual genome annotation process, we can use this dense information storage at runtime and thus achieve exact sequence identifications and ultra-fast lookups of related information.
We also reduced overall storage requirements to one third even though additional rich annotation information is included like gene symbols, EC numbers, GO terms, protein products and external database accessions. This information is a valuable resource to connect sequences at hand with related sequences stored in public databases.
Interestingly enough, this alignment-free approach also helped to substantially avoid computationally expensive alignments which follow as a fallback search strategy for unidentified sequences. In a hierarchical two-step process, remaining protein sequences were searched via traditional sequence alignments against protein cluster representative sequences. First, more than 99 million dense protein clusters were screened for matches followed by a second search using more-relaxed thresholds screening more than 13 million wider clusters.
Potentially negative runtime effects of these huge protein cluster databases were mitigated by the described alignment-free sequence identification approach. Finally, all annotation information for identified protein sequences and related clusters were combined giving specific information precedence over more general information.
This hierarchical approach is part of a larger annotation workflow also comprising the annotation of non-coding RNA and DNA features, e.g., tRNAs, rRNAs, ncRNAs, CRISPR arrays, origin of replications and many more. Bakta is available as a command line tool and as a scalable web service at https://bakta.computational.bio

This story is part of Science X Dialog, where researchers can report findings from their published research articles. Visit this page for information about ScienceX Dialog and how to participate.
More information:
Oliver Schwengers et al, Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification, Microbial Genomics (2021). DOI: 10.1099/mgen.0.000685
Oliver Schwengers is a microbial bioinformatics PostDoc researcher at the Bioinformatics and Systems Biology department at the JLU Giessen. His research activities focus on the analysis and characterization of bacterial genomes and plasmids based on whole-genome sequencing data as well as the development of fully automated and scalable bioinformatics software tools. He loves to regularly collaborate with researchers from medical, environmental and space microbiology in an interdisciplinary manner.
Citation:
Hashing complements alignment-based methods for bacterial genome annotation (2022, December 13)

