How do cells work in a normal state? How do they change when they cause disease? Do they react as desired to new drugs? Nowadays, anyone seeking answers to these—and other related—questions in the laboratory can hardly do without a special technique: single-cell RNA sequencing, or “scRNA-seq” for short. This technique provides an accurate picture of gene expression in a single cell at a specific point of time, as well as the associated regulatory networks, allowing conclusions to be drawn about the molecular basis of cell activity.
A research team at the Julius-Maximilians-Universität Würzburg (JMU) has now further improved a single-cell RNA sequencing technique it previously developed for use in bacteria. This means that the work in the laboratory is even faster than before and provides much more precise information. The team presents its development in the journal mBio.
High throughput thanks to automation
The leader of the study now published is Professor Jörg Vogel. Vogel heads the Institute for Molecular Infection Biology (IMIB) at JMU and is also director of the Helmholtz Institute for RNA-based Infection Research (HIRI). He is one of the world’s leading experts in the field of RNA research.
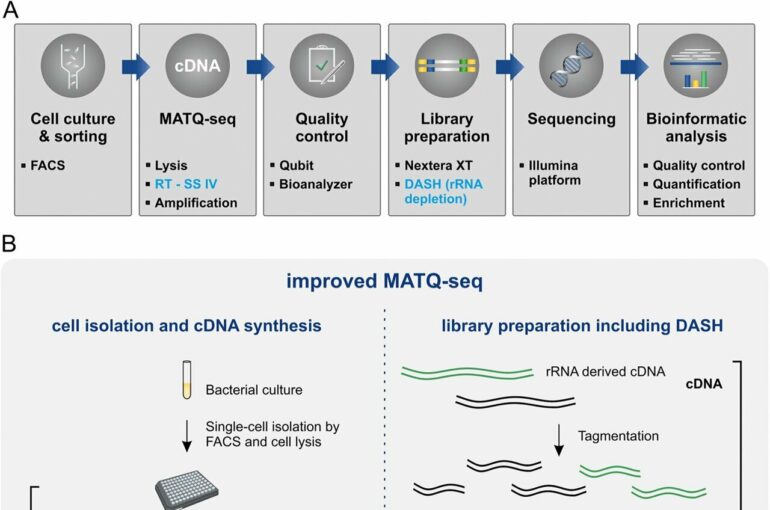
“By integrating a user-friendly and highly flexible automation process, we have achieved higher cell throughput,” says Vogel, describing one advantage of the method now being presented. In addition, the technique works more robustly, reducing the failure rate in reading genetic information, and provides more information about the gene expression of individual cells at lower sequencing costs.
Averages hide important details
Until a few years ago, studies of the transcriptome—the set of all genes active in a cell at a given time—in bacteria relied on bulk RNA sequencing (RNA-seq). “However, this approach only provides average values of a cell population and therefore does not allow any conclusions to be drawn about possible differences between individual bacteria within this population,” Vogel explains.
Such differences—in this case, scientists refer to them as “phenotypic heterogeneity”—are, however, often found in bacteria. They enable them to adapt quickly to changing environments and therefore assume an important role in bacterial survival strategies.
Bacteria pose special challenges for technology
While single-cell RNA sequencing was introduced in 2009 for eukaryotes—cells that have a nucleus—the development of this technique for bacteria has been much slower. A number of challenges are responsible for this: “Prokaryotic cells are much smaller compared to eukaryotes, which means you have much less material to study per cell,” Vogel explains. Other problems include breaking down the cell wall—known as cell lysis—and detecting specific bacterial transcripts.
It is true that bacterial single-cell transcriptomics has also recently become a reality thanks to technical advances. Nevertheless, there is room for improvement, for example, because the frequency of cell loss is too high or short transcripts, such as regulatory small RNAs (sRNAs), are poorly detected or cannot be measured at all. “In addition, transcript recognition is currently limited to about 200 genes per cell, which is far below the average bacterial transcriptome,” Vogel explains.
Successful validation on Salmonella
Some of these problems can be solved by the improvements in scRNA sequencing technology that have now been presented, as the research team was able to show by studying Salmonella-type bacteria under different growth conditions. The data show that the implemented changes increased cell throughput and the robustness of the protocol while reducing cell loss.
In addition, the scientists were able to improve gene coverage and gene detection limits. “We were even able to detect sRNAs at the single cell level, which had not been possible before,” Vogel says. This, he adds, will allow exploration of the regulatory functions of sRNA at the single-cell level in future studies.
In addition, the data confirm the heterogeneity within the same cell population, which could not be read from the average values of previous sequencing techniques. They now provide information, for example, on the activity of genes that are of particular importance for the disease-causing properties of these bacteria.
This makes the method particularly suitable for experiments where the starting material is limited, for example for the analysis of small subpopulations of bacterial cells in host niches or of intracellular bacteria.
More information:
Christina Homberger et al, Improved Bacterial Single-Cell RNA-Seq through Automated MATQ-Seq and Cas9-Based Removal of rRNA Reads, mBio (2023). DOI: 10.1128/mbio.03557-22
Provided by
University of Würzburg
Citation:
Improved RNA sequencing technologies provide deeper insights into bacteria (2023, March 8)

