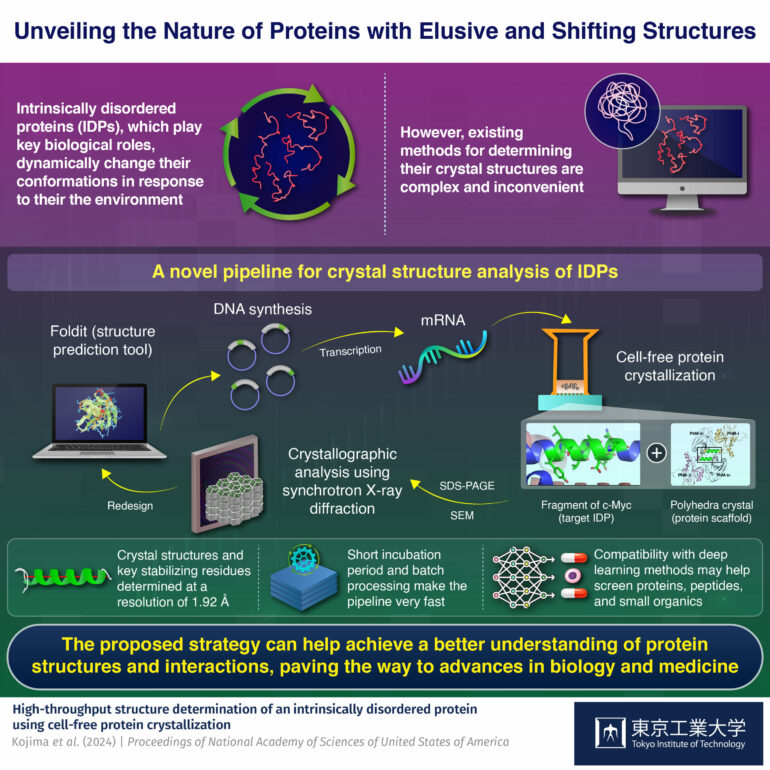
Intrinsically disordered proteins (IDPs) can dynamically change their conformations depending on their external environment and can, therefore, bind to different compounds. However, they are difficult to analyze. Now, Tokyo Tech researchers have addressed this issue with a novel pipeline that enables a rapid crystal structure analysis of IDPs via a cell-free protein crystallization technique.
Many people find it easier to think of proteins as some sort of rigid “molecular machinery,” with each protein having a well-defined structure that enables or complements its functions. However, many important proteins lack such a fixed three-dimensional structure. Instead, these so-called intrinsically disordered proteins (IDPs) can adopt a wide array of different conformations based on their external environment. This inherent flexibility of IDPs makes them versatile, and in general, capable of binding to many different compounds.
Compared to other types of proteins, IDPs can be quite difficult to analyze. To understand the biological functions of an IDP, it is useful to identify the factors—or determinants—that can stabilize its subregions at the atomic level. One prominent approach to achieve this is to immobilize a target IDP by having it bind to a protein crystal scaffold, which enables the use of protein crystallographic techniques. However, currently available methods to do this are quite slow and inconvenient to use.
Against this backdrop, a team of researchers led by Professor Takafumi Ueno from the School of Life Science and Technology and International Research Frontiers Initiative (IRFI), both at Tokyo Institute of Technology(Tokyo Tech), Japan, set out to establish a more reliable and versatile method. In their latest study, published in the Proceedings of the National Academy of Sciences, they report on the development of an innovative pipeline for the fast crystal structure analysis of IDPs.
One of the highlights of this pipeline is its use of a cell-free protein crystallization (CFPC) method to get a desired IDP to bind to a scaffold crystal. To illustrate the use of the pipeline, the researchers focused their efforts on a fragment of c-Myc, an IDP from a family of genes that regulate cell cycle progression, apoptosis, and other cellular functions.
Using Foldit, a protein structure prediction software, the team designed an insect cell-derived polyhedra crystal (PhC) as a scaffold onto which the c-Myc fragments would bind. To speed up the cross-crystallization between PhCs and c-Myc fragments, the researchers prepared c-Myc fused PhC monomer mRNA and mixed it in a system containing cell extracts and the building units of PhC.
Since the cell extracts contain the necessary cellular machinery to transcribe mRNA, this system can produce large quantities of c-Myc fused PhCs without relying on live cells, vastly accelerating the stabilization of the IDP and simplifying its subsequent extraction for analysis.
Once the crystallized c-Myc fragments were analyzed, the researchers used molecular dynamics simulation to strategically introduce mutations into the c-Myc gene before repeating the aforementioned steps. By comparing how the modified fragments bound to the PhC scaffold and the structures that formed, the team could determine the key residues that ultimately governed the stabilization of c-Myc.
“These findings underscore the power of our CFPC screening method as a valuable tool for determining the structures of challenging target proteins and elucidating the essential molecular interactions that govern their stability,” says Ueno.
The proposed strategy could be of great use in the study of biomolecular binding, which is a foundational aspect in fields like medicine and cell biology.

“Our screening system will be applied to target IDPs whose binding partners have not yet been identified and to design new binding molecules, such as inhibitors,” says Ueno. “Furthermore, the rapid screening of crystal structures could enable us to construct a design library of protein crystals, accelerating the elucidation of IDP folding mechanisms.”
More information:
Mariko Kojima et al, High-throughput structure determination of an intrinsically disordered protein using cell-free protein crystallization, Proceedings of the National Academy of Sciences (2024). DOI: 10.1073/pnas.2322452121
Provided by
Tokyo Institute of Technology
Citation:
New method enables fast crystal structure analysis of intrinsically disordered proteins (2024, June 13)

