Signals from the environment set off a cascade of changes that affect different genes in different ways. Therefore, traditionally, it has been difficult to study how such signals influence an organism. In a new study, researchers have developed a machine-learning approach called FUN-PROSE to predict how genes react to different environmental conditions.
Cells, regardless of the organism, fine-tune their reaction to their surroundings using mRNA. First, they use proteins called transcription factors that sense changes and then bind to the DNA sequence—called a promoter—in front of genes. This attachment can either stop the formation of mRNA from the gene or it can increase the amount of mRNA being made.
The mRNA then serves as a template to produce proteins responsible for various functions in the cell. This mechanism allows cells to rapidly reallocate resources to processes necessary for survival.
Studying how promoters are controlled is one of the oldest challenges in genomics, and yet researchers still continue to grapple with it. The biggest problem is that different transcription factors can bind to the same promoter sequence and do so in different arrangements under various environmental conditions.
Moreover, while there is some evidence that transcription factors tend to bind to specific sequence motifs in promoters, not all of them have been extensively studied. In recent years, researchers have turned to artificial intelligence to help them solve these challenges.
“Genes have an average level of expression, and previous machine learning models were unable to measure how the levels change under different conditions,” said Sergei Maslov (CAIM leader/CABBI), a professor of bioengineering and physics. “We were interested in understanding how specific genes react to changes in pH, temperature, and nutrients.”
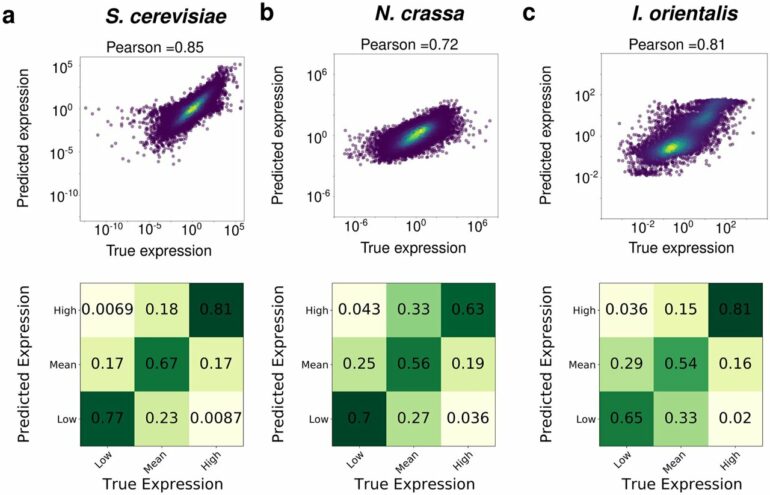
The researchers developed a model called FUNgal PRomoter to cOndition-Specific Expression, or FUN-PROSE, to predict how baker’s yeast (Saccharomyces cerevisiae) and the less studied fungi Neurospora crassa and Issatchenkia orientalis would react to environmental changes.
To develop the model, the researchers first had to identify promoter sequences and transcription factors for the three species. Then, they trained the model to learn what promoter motifs are recognized by transcription factors in different conditions.
“The transcription factors of N. crassa and I. orientalis are not as well-known as S. cerevisiae, so we had to infer what genes can be identified by transcription factors in these species,” said Ananthan Nambiar, a graduate student in the Maslov group. According to Veronika Dubinkina, a former graduate student in the Maslov group, now a postdoctoral researcher at Gladstone Institutes, this process involved a commonly used approach of scanning for protein regions that are known to bind DNA.
Finally, the model learned how to integrate all the information to calculate how much mRNA is made in a particular condition compared to the average level of mRNA. The researchers then compared the results obtained from FUN-PROSE to RNA-seq data, which measures fluctuating mRNA levels from all three fungi. Each organism has upwards of 4000 genes and 180 transcription factors that were measured in 12-295 conditions, depending on how well it has been studied.
“Predicting which genes are important under a set of conditions has always been a hard problem. However, we found that our model was very close to predicting what actually happens in these organisms,” Nambiar said.
In addition to evaluating its performance, the researchers elucidated how the model makes its predictions. “Even with its black-box nature, we were able to understand how our model looks at promoters and saw that it had learned to search for known sequences,” said Simon Liu, a former undergraduate in the Maslov group. “Being able to interpret the trained model is essential to validating its logic as well as using it to discover new regulatory knowledge.”
The model does, however, struggle with promoters that it hasn’t encountered before. “The model is great with novel conditions, but if you give it a novel gene or promoter sequence, it makes mistakes,” Nambiar said.
According to Maslov, these errors were due to the limited data available. “Machine learning is a black box, and you need to train it well so you can learn the biology,” he said. “If we can get more data, the model will have more patterns to learn from and will have more accurate predictions.”

The researchers are now interested in testing their model on other organisms. “In principle, there are no limitations to our technique—it should work on any organism. However, in animals, for example, genes are controlled in more complicated ways, which will require significant changes in the model architecture and much more training data,” Maslov said. “Still, it would be interesting to see how well this model does.”
The research is published in the journal PLOS Computational Biology.
More information:
Ananthan Nambiar et al, FUN-PROSE: A deep learning approach to predict condition-specific gene expression in fungi, PLOS Computational Biology (2023). DOI: 10.1371/journal.pcbi.1011563
Provided by
University of Illinois at Urbana-Champaign
Citation:
Predicting the response of fungal genes using a new machine-learning approach (2023, November 20)

