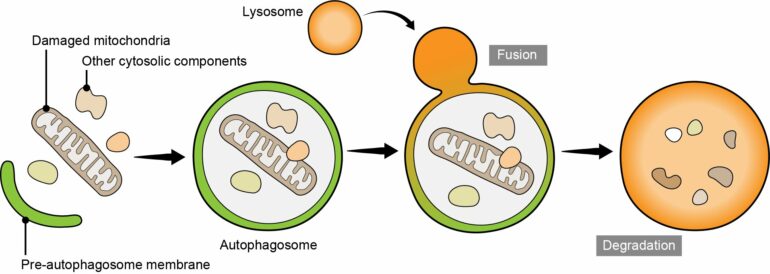
Autophagy is a process used by cells as a recycling system to transport and break down organelles and other cytosolic components, which become enveloped in a membrane called the autophagosome. When this involves the removal of damaged mitochondria, it is known as mitophagy.
In a recent article published in The EMBO Journal, a team led by researchers at Tokyo Medical and Dental University (TMDU) elucidated the molecular details of how an enzyme called Tank-binding kinase 1 (TBK1) participates in a disease-relevant mitophagy mechanism.
Although autophagy has been characterized as a more general process meant to degrade and clear various cellular components, recent data have suggested that certain pathways are specifically involved in the autophagy of particular organelle types that are damaged or no longer needed. The researchers became interested in mitophagy mediated by molecules called PINK1 and Parkin, as they are proteins that have been pathologically linked to Parkinson’s disease.
“Mitophagy-related defects have been directly implicated in the neurodegeneration observed in Parkinson’s disease patients,” says Koji Yamano, lead author of the study. “Normally, PINK1 and Parkin work together to mark damaged mitochondria for removal by adding a chain of molecules called ubiquitin. This mark allows proteins called autophagy adaptors to associate with the mitochondria and bring in the autophagy machinery for autophagosome development.”
 in wild-type cells, TBK1 gene deletion inhibits OPTN accumulation. © Department of Biomolecular Pathogenesis, TMDU")
While OPTN accumulates on the damaged mitochondria (at the mitophagy contact sites) in wild-type cells, TBK1 gene deletion inhibits OPTN accumulation. © Department of Biomolecular Pathogenesis, TMDU
Although TBK1 is known to participate in PINK1/Parkin-mediated mitophagy, a detailed mechanism of how it activates remained unclear. Using various molecular biology techniques, the team found that deleting the gene encoding TBK1 prevented the association of an autophagy adaptor called optineurin (OPTN) during Parkin-mediated mitophagy. Additionally, deleting the OPTN gene prevented autophosphorylation of TBK1, which is necessary for it to function.
Further work suggested that the interactions between OPTN and ubiquitin, as well as between OPTN and the developing autophagosome, were all needed for OPTN and TBK1 to come together at the contact site between damaged mitochondria and the pre-autophagosome membrane. Without this contact site, TBK1 autophosphorylation could not occur.
The researchers also generated molecules called monobodies in their lab that could specifically bind OPTN and inhibit its physical interactions. The monobodies prevented OPTN accumulation at the mitophagy contact sites. This subsequently blocked TBK1 activation and, thereby, mitochondrial degradation. These experiments further emphasized the importance of the OPTN-TBK1 relationship to support proper mitophagy.
 as well as TBK1 activation (B). © Department of Biomolecular Pathogenesis, TMDU")
Expression of GFP-Monobodies inhibits OPTN accumulation at the mitophagy contact sites (A) as well as TBK1 activation (B). © Department of Biomolecular Pathogenesis, TMDU
“Because PINK1 and Parkin are critical contributors to the molecular basis of Parkinson’s disease, understanding the mechanistic details related to the mitophagy process mediated by these molecules is very important,” explains Yamano.

This study demonstrates a positive and reciprocal relationship between OPTN and TBK1 that is necessary for autophagosomes to begin forming on damaged mitochondria. The impactful finding may lead to the development of novel drugs to treat Parkinson’s disease.
More information:
Koji Yamano et al, Optineurin provides a mitophagy contact site for TBK1 activation, The EMBO Journal (2024). DOI: 10.1038/s44318-024-00036-1
Provided by
Tokyo Medical and Dental University
Citation:
Research uncovers specific protein interactions needed for cells to break down and remove damaged mitochondria (2024, March 14)

