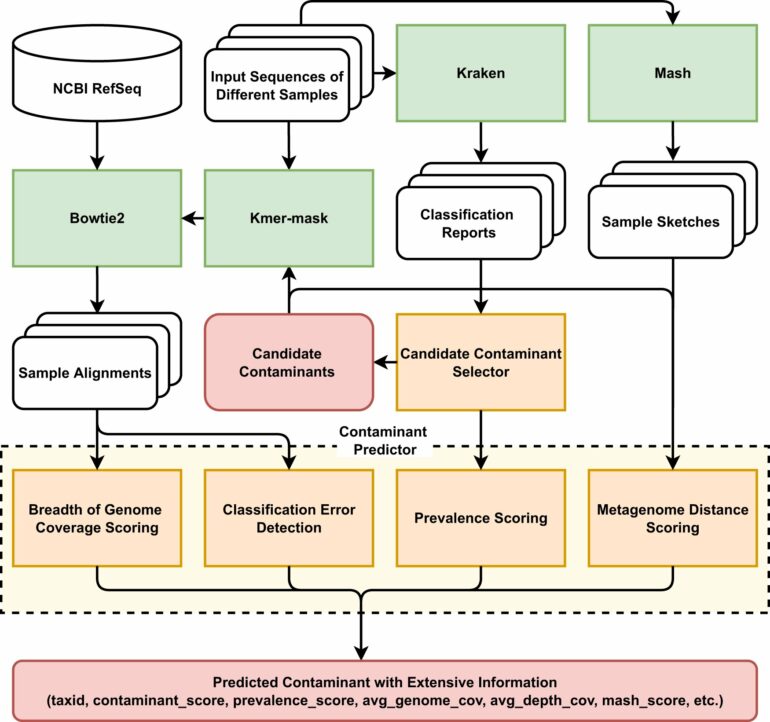
One of the major challenges in microbiome science has been distinguishing what is a potential environmental contaminant from a true, bona fide microbiome signal. Challenges associated with metagenomic sequencing with low biomass environments include the distinction between a true signal versus contamination, a remnant DNA from a sampling kit or extraction kit or the environment.
While researchers normally include negative controls from the equipment or environment and use algorithmic tools to identify taxa present in the environment, not all datasets come with negative controls. Researchers at Baylor College of Medicine and Rice University developed a de novo contamination detection tool to establish reproducibility in the identification and analysis of the microbes. Their findings were recently published in Nature Communications.
“We teamed up with our collaborators at Rice University to develop and test a computational tool we called Squeegee,” said Dr. Kjersti Aagaard, professor of obstetrics and gynecology at Baylor and Texas Children’s Hospital.
“The premise of Squeegee is that we can use computer analysis pipeline to help us detect ‘breadcrumbs’ of contaminants that would be anticipated to be common between the microbiome found in all human (or other mammalian) hosts and the sampling or lab environment.”
The Aagaard Lab at Baylor has conducted research over the last decade leading to a number of rich datasets from a large number of participants that are particularly low biomass and have many negative controls. They teamed up with researchers at Rice’s Treangen Lab to test Squeegee, an algorithm used on life datasets from human studies that had contamination controls from different environments and DNA extraction kits.
They looked at the false positive rate, the recall and how accurately Squeegee could predict and flag these environmental contamination sets with the absence of the negative control.
“We were able to show that Squeegee was capable of having a high-weighted recall and a very low false-positive rate in these ground truth datasets,” said Dr. Michael Jochum, postdoctoral research associate in the Department of Obstetrics and Gynecology Baylor.
According to Jochum, Squeegee improves the overall reliability of metagenomic sequencing analysis results in low biomass studies—studies that contain little microbial DNA like breastmilk, placenta or amniotic fluid. The de novo contamination identification tool is capable of identifying batch effects, flagging them as potential contaminants. Given the focus and expertise of the Aagaard lab in studying these sparse microbial environments, this is a tool that they have added to their toolbox for ongoing and future studies.
“This is a first-of-its-kind tool for the microbiome science community, and it is freely available for use,” Aagaard said.

More information:
Yunxi Liu et al, De novo identification of microbial contaminants in low microbial biomass microbiomes with Squeegee, Nature Communications (2022). DOI: 10.1038/s41467-022-34409-z
Provided by
Baylor College of Medicine
Citation:
Researchers develop algorithm to identify microbial contaminants in low microbial biomass microbiomes (2022, November 22)

