DeepMind, the AI unit of Google that invented the chess champ neural network AlphaZero a few years back, shocked the world again in November with a program that had solved a decades-old problem of how proteins fold. The program handily beat all competitors, in what one researcher called a “watershed moment” that promises to revolutionize biology.
AlphaFold 2, as it’s called, was described at the time only in brief terms, in a blog post by DeepMind and in a paper abstract provided by DeepMind for the competition in which they submitted the program, the Critical Assessment of Techniques for Protein Structure Prediction biannual competition.
Last week, DeepMind finally revealed just how it’s done, offering up not only a blog post but also a 16-page summary paper written by DeepMind’s John Jumper and colleagues in Nature magazine, a 62-page collection of supplementary material, and a code library on GitHub. A story on the new details by Nature’s Ewan Calloway characterizes the data dump as “protein structure coming to the masses.”
So, what have we learned? A few things. As the name suggests, this neural net is the successor to the first AlphaFold, which had also trounced competitors in the prior competition in 2018. The most immediate revelation of AlphaFold 2 is that making progress in artificial intelligence can require what’s called an architecture change.
The architecture of a software program is the particular set of operations used and the way they are combined. The first AlphaFold was made up of a convolutional neural network, or “CNN,” a classic neural network that has been the workhorse of many AI breakthroughs in the past decade, such as containing triumphs in the ImageNet computer vision contest.
But convolutions are out, and graphs are in. Or, more specifically, the combination of graph networks with what’s called attention.
A graph network is when some collection of things can be assessed in terms of their relatedness and how they’re related via friendships — such as people in a social network. In this case, AlphaFold uses information about proteins to construct a graph of how near to one another different amino acids are.
Also: Google DeepMind’s effort on COVID-19 coronavirus rests on the shoulders of giants
These graphs are manipulated by the attention mechanism that has been gaining in popularity in many quarters of AI. Broadly speaking, attention is the practice of adding extra computing power to some pieces of input data. Programs that exploit attention have lead to breakthroughs in a variety of areas, but especially natural language processing, as in the case of Google’s Transformer.
The part that used convolutions in the first AlphaFold has been dropped in Alpha Fold 2, replaced by a whole slew of attention mechanisms.
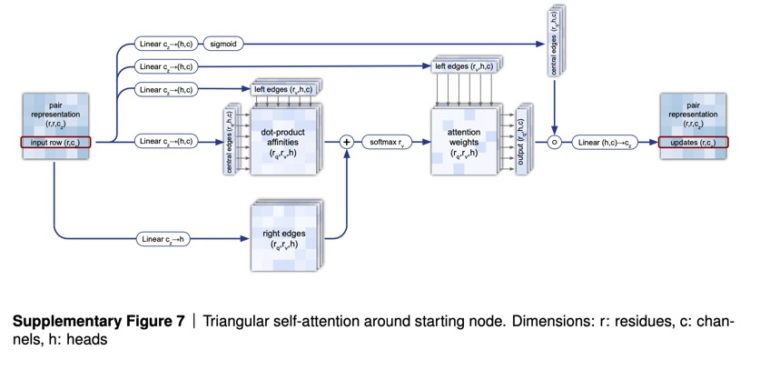
Use of attention runs throughout AlphaFold 2. The first part of AlphaFold is what’s called EvoFormer, and it uses attention to focus processing on computing the graph of how each amino acid relates to another amino acid. Because of the geometric forms created in the graph, Jumper and colleagues refer to this operation of estimating the graph as “triangle self-attention.”
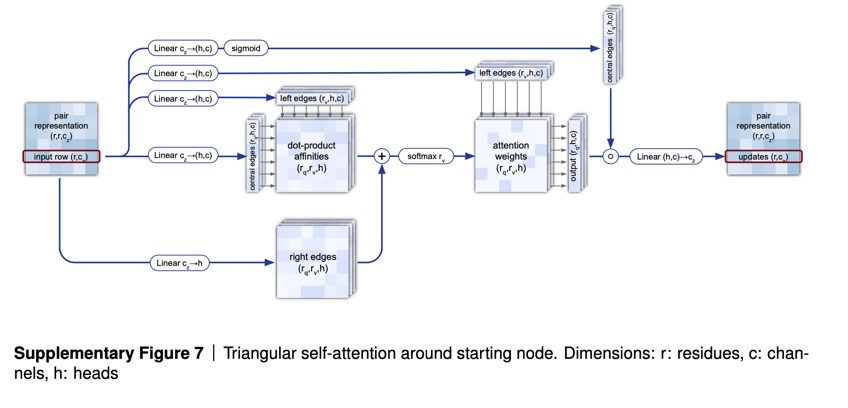
Echoing natural language programs, the EvoFormer allows the triangle attention to send information backward to the groups of amino acid sequences, known as “multi-sequence alignments,” or “MSAs,” a common term in bioinformatics in which related amino acid sequences are compared piece by piece.
The authors consider the MSAs and the graphs to be in a kind of conversation thanks to attention — what they refer to as a “joint embedding.” Hence, attention is leading to communication between parts of the program.
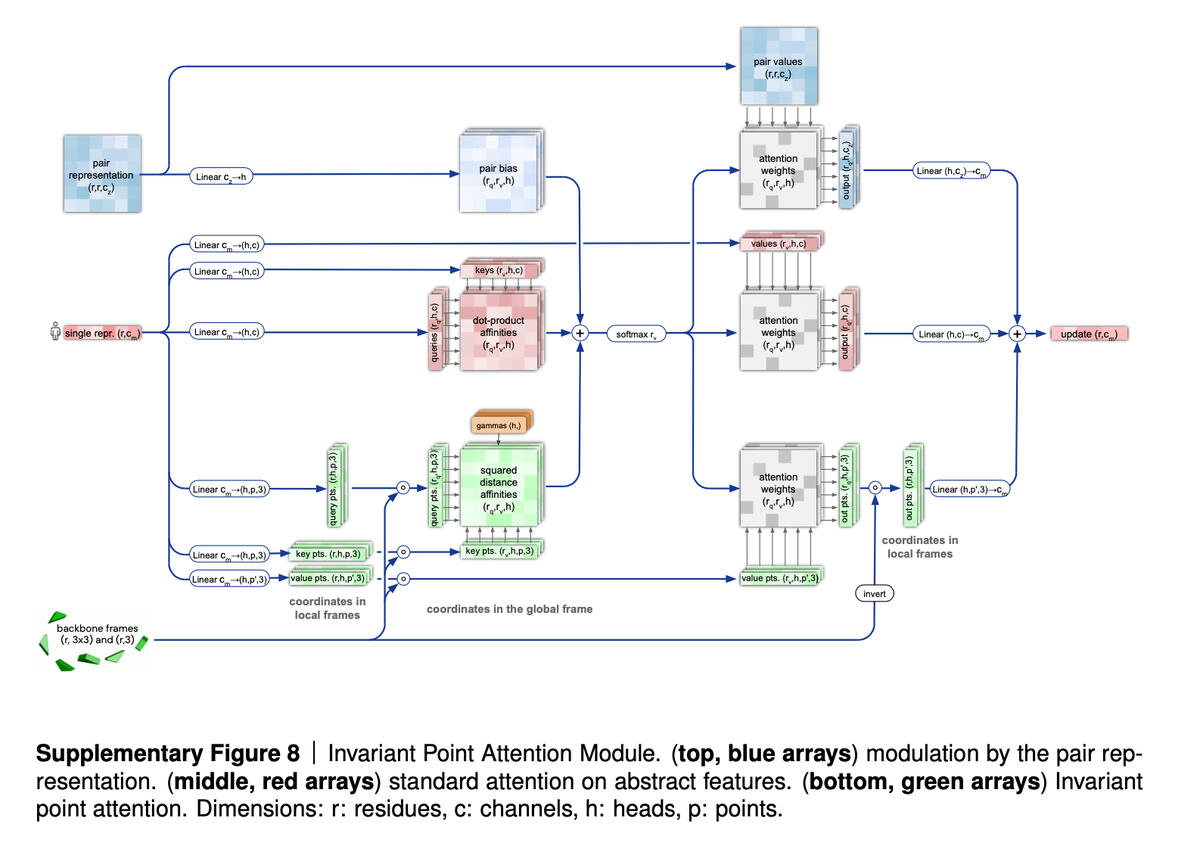
The second part of AlphaFold 2, following the EvoFormer, is what’s called a Structure Module, which is supposed to take the graphs that the EvoFormer has built and turn them into specifications of the 3-D structure of the protein, the output that wins the CASP competition.
Here, the authors have introduced an attention mechanism that calculates parts of a protein in isolation, called an “invariant point attention” mechanism. They describe it as “a geometry-aware attention operation.”
The Structure Module initiates particles at a kind of origin point in space, which you can think of as a 3-D reference field, called a “residue gas,” and then proceeds to rotate and shift the particles to produce the final 3-D configuration. Again, the important thing is that the particles are transformed independently of one another, using the attention mechanism.
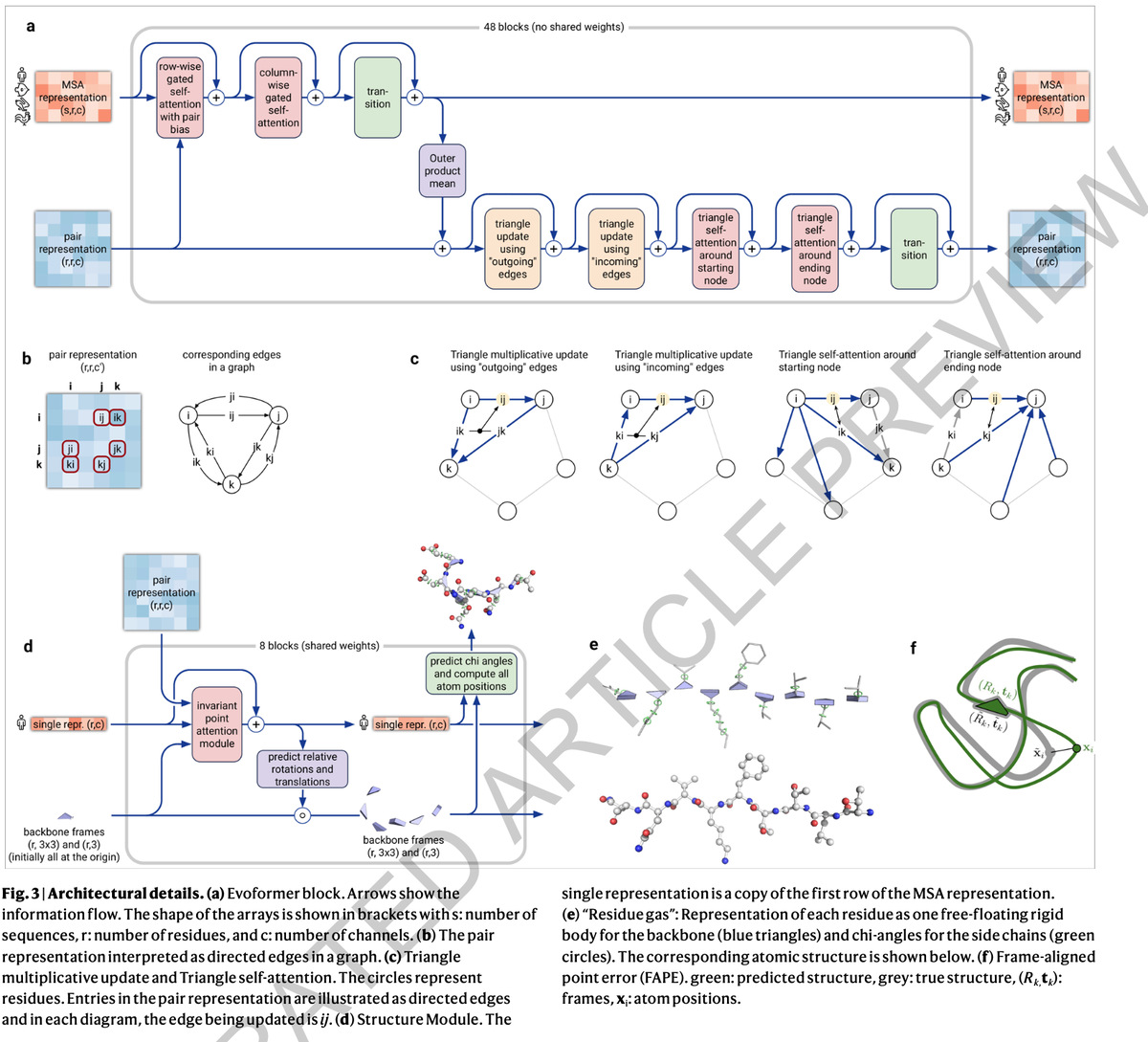
Why is it important that graphs, and attention, have replaced convolutions? In the original abstract offered for the research last year, Jumper and colleagues pointed out a need to move beyond a fixation on what are called “local” structures.
Going back to AlphaFold 1, the convolutional neural network functioned by measuring the distance between amino acids, and then summarizing those measurements for all pairs of amino acids as a 2-D picture, known as a distance histogram, or “distogram.” The CNN then operated by poring over that picture, the way CNNs do, to find local motifs that build into broader and broader motifs spanning the range of distances.
But that orderly progression from local motifs can ignore long-range dependencies, which are one of the important elements that attention supposedly captures. For example, the attention mechanism in the EvoFormer can connect what is learned in the triangle attention mechanism to what is learned in the search of the MSA — not just one section of the MSA, but the entire universe of related amino acid sequences.
Hence, attention allows for making leaps that are more “global” in nature.
Another thing we see in AlphaFold is the end-to-end goal. In the original AlphaFold, the final assembly of the physical structure was simply driven by the convolutions, and what they came up with.
In AlphaFold 2, Jumper and colleagues have emphasized training the neural network from “end to end.” As they say:

“Both within the Structure Module and throughout the whole network, we reinforce the notion of iterative refinement by repeatedly applying the final loss to outputs then feeding the outputs recursively to the same modules. The iterative refinement using the whole network (that we term ‘recycling’ and is related to approaches in computer vision) contributes significantly to accuracy with minor extra training time.”
Hence, another big takeaway from AlphaFold 2 is the notion that a neural network really needs to be constantly revamping its predictions. That is true both for the recycling operation, but also in other respects. For example, the EvoFormer, the thing that makes the graphs of amino acids, revises those graphs at each of the multiple stages, what are called “blocks,” of the EvoFormer. Jumper and team refer to this constant updates as “constant communication” throughout the network.
As the authors note, through constant revision, the Structure piece of the program seems to “smoothly” refine its models of the proteins. “AlphaFold makes constant incremental improvements to the structure until it can no longer improve,” they write. Sometimes, that process is “greedy,” meaning, the Structure Module hits on a good solution early in its layers of processing; sometimes, it takes longer.
Also: AI in sixty seconds
In any event, in this case the benefits of training a neural network — or a combination of networks — seem certain to be a point of emphasis for many researchers.
Alongside that big lesson, there is an important mystery that remains at the center of AlphaFold 2: Why?
Why is it that proteins fold in the ways they do? AlphaFold 2 has unlocked the prospect of every protein in the universe having its structure revealed, which is, again, an achievement decades in the making. But AlphaFold 2 doesn’t explain why proteins assume the shape that they do.
Proteins are amino acids, and the forces that make them curl up into a given shape are fairly straightforward — things like certain amino acids being attracted or repelled by positive or negative charges, and some amino acids being “hydrophobic,” meaning, they stay farther away from water molecules.
What is still lacking is an explanation of why it should be that certain amino acids take on shapes that are so hard to predict.
AlphaFold 2 is a stunning achievement in terms of building a machine to transform sequence data into protein models, but we may have to wait for further study of the program itself to know what it is telling us about the big picture of protein behavior.

