Vaccines to block COVID-19 that are in development by Moderna, Pfizer, AstraZeneca and others, and that are currently in Phase III clinical trials, may not do as well covering people of Black or Asian genetic ancestry as they do for white people, a study released Wednesday by the Massachusetts Institute of Technology indicated.
“There are obviously many other factors to consider, but our preliminary results suggest that, on average, people of Black or Asian ancestry could have a slightly increased risk of vaccine ineffectiveness,” one of the authors of the report, David K. Gifford, who is with MIT’s Computer Science and Artificial Intelligence Laboratory (CSAIL), said in a press release issued by MIT.
The report, entitled “Predicted Cellular Immunity Population Coverage Gaps for SARS-CoV-2 Subunit Vaccines and their Augmentation by Compact Peptide Sets,” was posted on the Bioarxiv pre-print server and has not yet been peer-reviewed, which means its findings must be taken with an extra degree of caution.
Enthusiasm has surged in recent weeks as Moderna, Pfizer and AstraZeneca all announced initial results from Phase III trials in human patients that showed surprisingly powerful rates of immunity, with tests subjects given the drugs being 94% to 95% less likely than people given a placebo to contract COVID-19.
Those three vaccine efforts are only the most prominent in a vast array of efforts. There are fifty-one vaccines in clinical trials in total, according to the World Health Organization. There are another one hundred and sixty-three vaccines in a pre-clinical stage of evaluation.
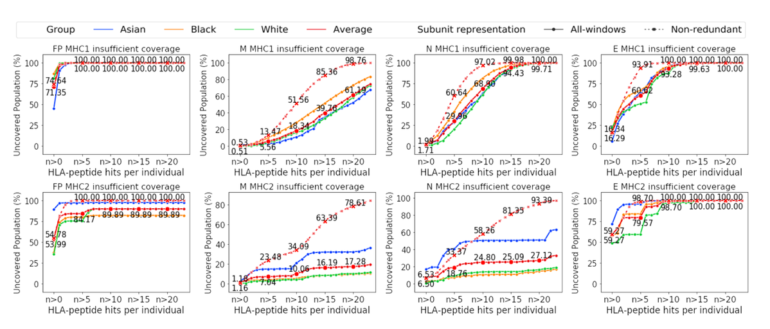
Curves of coverage show varying percentages of people by genetic ancestry that are likely to gain an immune response to COVID-19, for different mixes of alleles and viral peptide combinations.
Liu et al
Many of the vaccines, including those from Moderna and Pfizer and AstraZeneca, share the same weakness, the MIT report contends, which is that they do not use a sufficiently diverse set of viral particles to stimulate the same level of immune response in all people in the population, depending on genetic makeup.
The report draws on in silico computer models. Gifford and co-authors Ge Liu and Brandon Carter, two PhD students with MIT’s CSAIL, used machine learning models to predict, based on patient data and models of proteins in the immune system, how likely vaccines would be to have a “hit,” meaning, to successfully stimulate an immune response, in different population groups based on self-reported ethnic type or genetic ancestry.
Also: MIT’s machine learning designed a COVID-19 vaccine that could cover a lot more people
The work in the paper builds on work done this summer by the group to develop two computer models that predict vaccine coverage. One, called OptiVax, predicts a vaccine’s stimulation of immune responses. A second, called EvalVax, maps that immune response to the biochemistry of population groups by ethnic or genetic ancestral status.
The vaccine mechanism is modeled by the programs. When an invading organism enters the body, such as a virus, some of the bits of virus, short strings of perhaps 8 to 25 amino acids, known as peptides, fit into a groove in the surface of a person’s cells. The cell is then able to present the bits of the virus to the body’s T cells as a signal of the invasion. The T cells begin a process of killing off such infected cells.
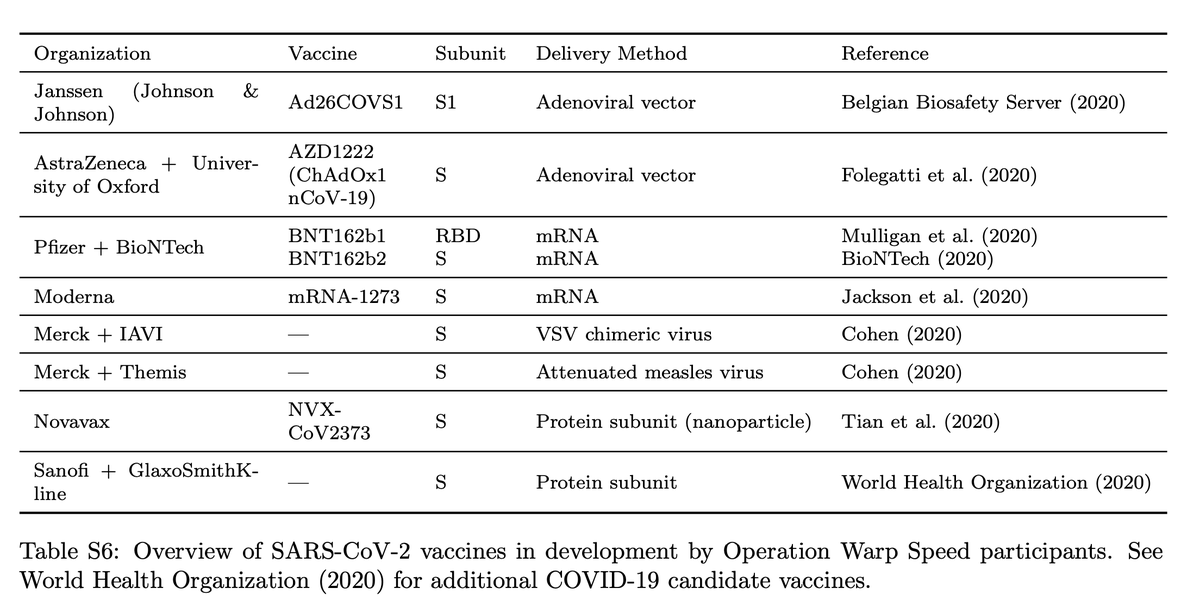
Vaccines in development. Gifford and colleagues contend all drugs in the U.S. Operation Warp Speed program, including those from Moderna and Pfizer and AstraZeneca, have the same weakness, which is that they cluster around a vary narrow selection of viral peptides to stimulate immune response, which deprives them of sufficient diversity to ensure the broadest population coverage.
Liu et al
That’s how natural human immunity works, and vaccines mimic that process by using a bit of the virus specially engineered to artificially simulate the cell’s response.
In the report this summer, Gifford and team had warned that not using enough different parts of the virus could leave gaps in coverage. That is because humans have different “alleles,” versions of genes, in what’s called the major histocompatibility complex, the area of the human genome that encodes the cell-surface receptors that are supposed to match the viral peptides. Some alleles produce cell receptors that will bind more or less reliably to some viral peptides.
In the present study, Gifford and team built upon that study to show that the vaccines from Moderna and Pfizer and AstraZeneca have exactly the weakness that the researchers had predicted in their computer modeling.
Also: MIT’s deep learning found an antibiotic for a germ nothing else could kill
All the vaccines are using the same bits of the virus, the so-called Spike protein, or S protein, and a special area of the Spike protein, called the Receptor Binding Domain, or RBD. “All reported current efforts for COVID-19 vaccine design that are part of the United States Government’s Operation Warp Speed use variants of the spike subunit of SARS-CoV-2 to induce immune memory,” Gifford and team write.
That focus on a limited number of viral peptides becomes a common weakness in the vaccines, they argue. “We find that proposed SARS-CoV-2 subunit vaccines exhibit population coverage gaps in their ability to generate a robust number of predicted peptide-HLA hits in every individual.” HLAs is the technical term for the cell surface receptors that bind with the peptides.

The researchers relate several instances where their models show that the lack of greater peptide diversity leads to widely varying coverage:
Based on our prediction, the receptor binding domain (RBD) subunit had no MHC class II peptides displayed in 15.12% of the population (averaged across Asian, Black, and White self-reporting individuals) […] We note that the uncovered population of RBD with no predicted display of MHC class II peptides ranges from 0.811% for the popu- lation self-reporting as White, to a high of 37.287% for the population self-reporting as Asian.
ZDNet reached out to Moderna, Pfizer, and AstrZeneca for comment and will update the article with any response.
The authors have a couple of suggestions for the drug makers. One is to take into account genetic ancestry explicitly. “Clinical trials need to carefully consider ancestry in their study designs to ensure that efficacy is measured across an appropriate population,” they write.
Second, as they did in de novo vaccine synthesis over the summer, Gifford and collaborators were able to tweak vaccine designs to include a greater diversity of peptides.
Their computer model of the drug designs suggests the proportion of people who would be covered would substantially improve if a greater mix of peptides is included, the authors write:
The computed sets of augmentation peptides were predicted to substantially reduce the populations predicted to be insufficiently covered by each subunit. Post augmentation the predicted uncovered population for RBD with no peptide-MHC hits is reduced to 0.003% (MHC class I) and 4.351% (MHC class II) with MIRA positive peptides only, and 0.0% (MHC class I) and 0.309% (MHC class II) with all filtered peptides from SARS-CoV-2. (Table S1).
The authors note that their code and data is freely available on GitHub.

