In a continuing effort to improve upon previous work, a research team at the Graduate School of Science, Osaka City University, has applied its recently developed Bayesian phase difference estimation quantum algorithm to perform full configuration interaction (full-CI) calculations of atoms and molecules without simulating the time evolution of the wave function conditional on an ancillary qubit. Superior to conventional methods in terms of parallel execution of quantum gates during quantum computing, this new algorithm is expected to be much easier to implement in actual quantum computers.
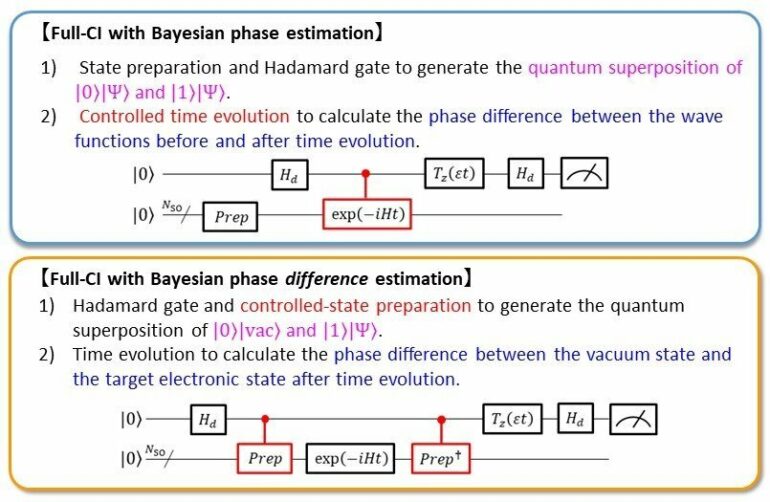
The researchers at the Osaka City University Graduate School of Science continue to improve their recently developed quantum algorithm, this time by applying it to calculate the potential energy curves of the H2 molecule without the need for controlled time evolutions. By including a controlled-state preparatory step to the Bayesian phase difference estimation (BPDE) quantum algorithm they previously developed, the team calculated the superposition of the “vacuum” wave function with zero electrons and the wave function of the desired electronic state, bypassing the need to simulate the time evolution of the wave function conditional on an ancillary qubit. This addresses an issue common to conventional quantum algorithms, namely the parallel processing of quantum gates and high number of quantum gates between two non-neighboring qubits—demonstrating itself as a quantum algorithm executable on quantum computers that can perform full configuration interaction (full-CI) calculations of atoms and molecules. Note that the full-CI calculations are capable of giving the optimized solutions of Schrödinger equations for microscopic systems, but not intractable for the sizable systems with classical computers because of the exponential explosion of required computing time.
Their research was published in The Journal of Physical Chemistry Letters as an open access article.
In the race to accurately solve the Schrödinger equation to show the electronic state of an atom or molecule, bringing a paradigm shift in scientific research and materials development, scientists have turned to quantum computers to carry out chemical calculations in polynomial time. The quantum phase estimation (QPE) algorithm has become a well-known and powerful tool in giving full-CI calculations of wave functions for small molecules, and various attempts have been made to have QPE-based methods account for the computational costs that exponentially accumulate against the system size under study.
“QPE-based methods simulate the time evolution of a wave function on an ancillary qubit, which requires many controlled quantum gates, the presence of which also hinders parallel execution of these gates and compression of quantum circuits,” states lead author, Specially-Appointed Lecturer Kenji Sugisaki. “In this research, we have applied our Bayesian phase difference estimation (BPDE) algorithm, which is a modification of QPE and capable of bypassing the need of controlled time evolution operations, to perform full-CI calculations. We emphasize that our approach invokes the energy calculation of many-electron systems as referring to the counterpart of many-electron ionized systems.”
“Evolving the wave function only when the first qubit is in the |1⟩ state makes it difficult to parallelize the quantum gates,” says research advisor, Professor Kazunobu Sato. “Rewriting the quantum logic circuit so that the time evolution operator can be applied regardless of whether the first qubit is in the |0⟩ or |1⟩ state would allow easy parallel processing of quantum gates and increase the likelihood of implementing the algorithm in actual quantum computers.”
To do this, the team introduced a controlled-state preparation that constructs the quantum superposition of the “vacuum” wave function |vac⟩ with zero electrons and the wave function |Ψ⟩ of the target electronic state. “In other words, we computed the full-CI energy of an atom or molecule as its ionization energy,” explains Professor Emeritus Takeji Takui, with the following logic circuit (|0⟩|vac⟩+|1⟩|Ψ⟩)⁄√2. The team exemplified the efficiency of their BPDE-based full-CI method by calculating the potential energy curves of four valence electronic states of the H2 molecule. “The numerical simulations revealed that the BPDE-based method reproduces the full-CI energy within 3 kcal mol-1 of errors for all the electronic states,” continues Prof. Takui, who also served as an advisor to the study.
Importantly, being free from controlled time evolution makes parallelization of quantum gates and implementation on real quantum devices easier, which gives the team hope that their Bayesian phase difference estimation algorithm paves the way for more practical full-CI calculations and becomes synonymous with precise quantum chemistry.

More information:
Kenji Sugisaki et al, Quantum Algorithm for Full Configuration Interaction Calculations without Controlled Time Evolutions, The Journal of Physical Chemistry Letters (2021). DOI: 10.1021/acs.jpclett.1c03214
Provided by
Osaka City University
Citation:
Newly improved quantum algorithm performs full configuration interaction calculations without controlled time evolutions (2021, November 26)

