Scientists studying a COVID-19 coronavirus enzyme at temperatures ranging from frosty to human-body warm discovered subtle structural shifts that offer clues about how the enzyme works. The findings, published in IUCrJ, the journal of the International Union of Crystallography, may inspire the design of new drugs to counteract COVID-19—and possibly help head off future coronavirus pandemics.
“No previous study has looked at this important coronavirus enzyme at physiological (or body) temperature,” said Daniel Keedy, a structural biologist at the City University of New York (CUNY), who conducted the study in collaboration with scientists at the U.S. Department of Energy’s Brookhaven National Laboratory.
Most structures to date come from frozen samples—far from the temperatures at which the molecules operate within living cells. “If you are working at physiological temperature, you should get a more realistic picture of what’s happening during an actual infection, because that’s where biology happens,” Keedy said.
In addition, he added, the team used temperature as a tool. “By turning that knob and seeing how the protein reacts, we can learn about its mechanics—how it physically works.
Deciphering Mpro’s structure
The protein in question is the main protease (Mpro) of SARS-CoV-2, the virus that causes COVID-19. Like all proteases, it’s an enzyme that cuts other proteins. In many viral infections, including COVID-19, infected cells initially produce a virus’s functional proteins as one single connected protein chain. Proteases cut the pieces apart so the individual proteins can make and assemble themselves into new copies of the virus. Finding a drug to disable Mpro could put the brakes on COVID-19.
To study the enzyme’s structure, the researchers used a technique called X-ray crystallography at Brookhaven Lab’s National Synchrotron Light Source II (NSLS-II). NSLS-II is a DOE Office of Science user facility that produces bright beams of x-rays. Shining those x-rays on a crystallized sample of a biological molecule can reveal the three-dimensional arrangement of atoms that make up the molecule.
Studying samples that aren’t frozen can be a challenge.
“The higher the temperature, the greater the chances the x-rays will damage the crystal,” explained study co-author Babak Andi, who operates NSLS-II’s Frontier Macromolecular Crystallography (FMX) beamline.
“To minimize the damage, we rotate and move the crystal linearly as it moves through the x-rays. That distributes the X-ray dose over the whole length of the crystal,” he said.
He noted that the small size of the X-ray beam at NSLS-II makes it possible to keep the beam focused on even the smallest dimension of the crystal—an edge measuring 10 to 20 millionths of a meter or smaller—as it rotates.
“In addition, the FMX detector and other systems operate so fast, we can collect one full dataset in just 10-15 seconds per sample, with good enough quality to solve a structure before significant X-ray damage occurs.”
From frozen to physiological
The scientists used FMX to collect the first-ever series of Mpro crystallographic data at five different temperatures, ranging from cryogenic (-280 degrees Fahrenheit) to what is often referred to as “room temperature” in X-ray crystallography (~39°F) to physiological (98°F). They also studied the room-temperature crystal under high humidity. Then they fed the data into a special type of computer simulation to identify many possible atomic-level arrangements under each set of conditions.
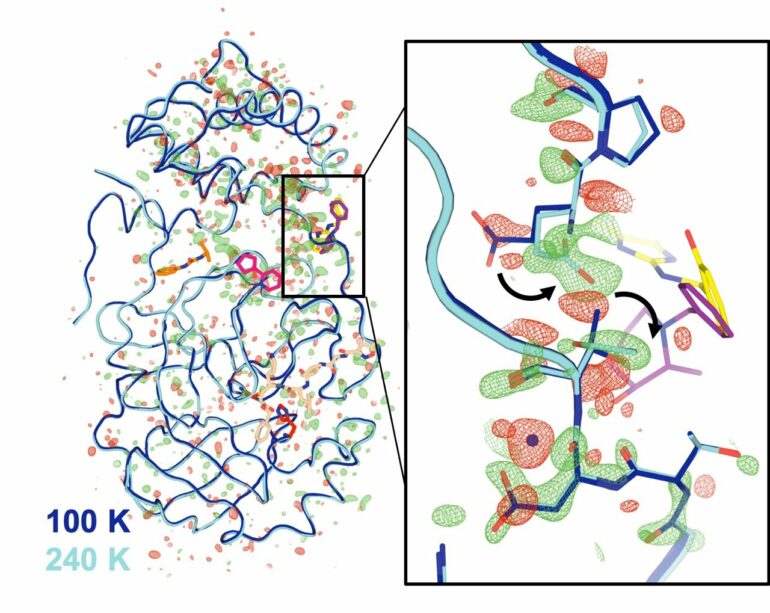
The results revealed subtle conformational changes, including increased flexibility in parts of the protein at higher temperatures. The team also saw some features that were unique to the enzyme under physiological conditions.
 but different conditions of relative humidity (RH). A comparison reveals one extra water molecule (H2O) near the active site under the high humidity condition (b) that was not present at ambient humidity (a). This water molecule could offer a clue where an inhibitor molecule might bind to disable the Mpro enzyme. © Brookhaven National Laboratory/CUNY")
Structures of the Mpro active site taken at the same temperature (298 Kelvin, K, or ~77°F) but different conditions of relative humidity (RH). A comparison reveals one extra water molecule (H2O) near the active site under the high humidity condition (b) that was not present at ambient humidity (a). This water molecule could offer a clue where an inhibitor molecule might bind to disable the Mpro enzyme. © Brookhaven National Laboratory/CUNY
Most of the changes were not directly in the enzyme’s “active site”—the part that is directly involved in cutting other proteins. Instead, they were in parts of the enzyme more distant from that location. But the data suggest that these distant sites could affect the active site through a sort of remote-control mechanism that’s common in biological systems, Keedy said. Disabling even those distant locations could potentially block the enzyme’s function.
“You can think of Mpro as a kind of folded ribbon, made up of two identical halves (forming dimers) that bind together in a symmetrical way, kind of like a handshake,” Keedy said. The center of this handshake region (the “dimer interface”) links to the active site through a flexible loop region of the protein.
As Keedy explained, the scientists found that at higher temperatures, “the grip of the ‘handshake’ changes—the two components readjust their grip a little bit. This tells us that, when the virus is infecting us, there may be some kind of communication via this loop between the dimer interface and the active site,” Keedy said.
The path to drug design
“We see subtle changes in structure in this study, but drug design is dependent on subtle changes—less than a billionth of a meter here, less than a billionth of a meter there,” Keedy noted.
Other studies have shown that small drug-like molecules can bind to the enzyme at some of distant locations identified in this new work.
“If we could perfect these molecules, optimize them, elaborate on them, tweak them, then we could potentially have a new foothold for altering the function of the enzyme—not at the active site, as essentially all of the antivirals for this protein are currently targeting, but at a different site through a different mechanism,” Keedy said. “Our findings set up the inspiration for exploring this idea.”
The role of water
Exploring the enzyme at elevated humidity also mimics the physiological conditions within water-filled cells—and may provide additional clues to guide drug design.
“For these studies, after selecting the crystal we want to study, we put a special sleeve over it to keep it from drying out,” said NSLS-II’s Babak Andi. “Then, when we put the sample in the beamline for X-ray data collection, we remove the sleeve and blow a continuous stream of air at 99.5% humidity over the crystal as we collect the data.”
The results revealed specific water molecules that bind loosely to the enzyme, including one near the active site, only under the high-humidity condition.
“Those water molecules give you a hint where inhibitors may bind,” said Andi.
His group has been studying a range of potential drug-like molecules that appear to replace weakly bound water molecules when they bind near Mpro’s active site. That work is reported on in a paper recently published in Scientific Reports from the Nature portfolio of journals.
Both scientists were grateful for the collaborative spirit of the entire team, which included additional scientists at both Brookhaven Lab and CUNY, as well as Diamond Light Source in the United Kingdom.
“It was very important to be able to have the remote collaboration to make this project work, where we had people onsite at the beamline, and people offsite who could do the computer modeling,” Keedy said. “It’s one example of many during the pandemic of the scientific community coming together.”
More information:
Ali Ebrahim et al, The temperature-dependent conformational ensemble of SARS-CoV-2 main protease (Mpro), IUCrJ (2022). DOI: 10.1107/S2052252522007497
Provided by
Brookhaven National Laboratory
Citation:
First structure of key COVID enzyme at human body temperature (2022, August 17)

