Researchers at the University of Toronto have discovered how mitochondrial turnover—a critical cellular function—begins.
Mitochondria are like the batteries of our bodies. They’re vital sources of energy for cells and are necessary to regulate function in almost all cell types. And, like batteries, mitochondria need to be replaced as they run down over time. If these cell batteries aren’t replaced efficiently, and don’t turn over properly, cells experience stress and can die.
Healthy mitochondria, in turn, are critical in energy-demanding organs such as the brain and muscle. When this degradation process is disrupted, vulnerable neurons can die. This type of disruption is present in many neurodegenerative diseases such as Parkinson’s.
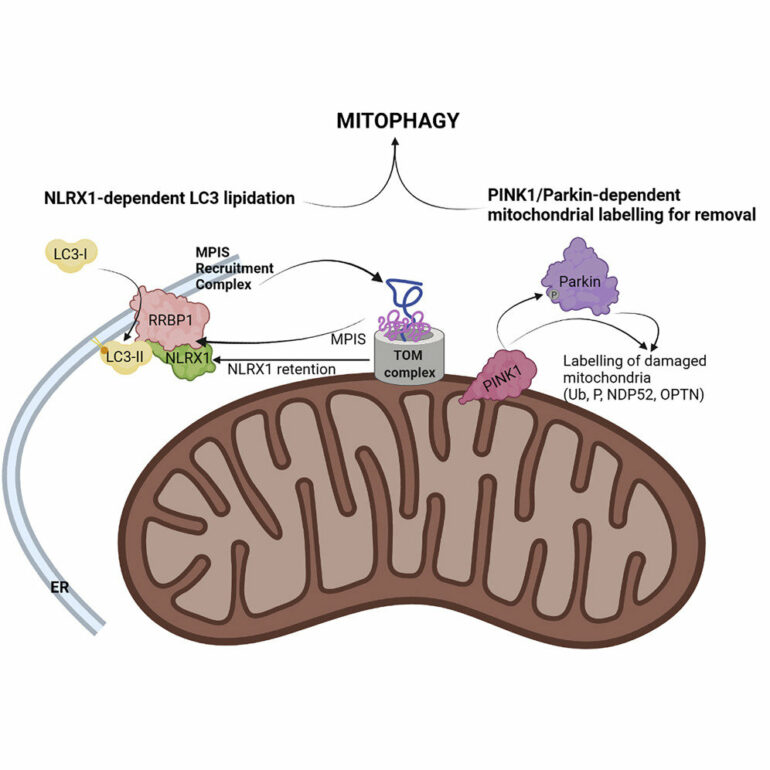
Now, a study by Stephen Girardin, a professor of laboratory medicine and pathobiology in the Temerty Faculty of Medicine, and post-doctoral researcher Samuel Killackey shows that when certain nuclear encoded proteins aren’t brought into mitochondria, the mitochondria are removed.
“We’re proud we’ve identified the problem and made progress toward understanding and characterizing the molecular players and pathways, and how this is all integrated in a cell—in some surprising ways,” Giradin says.
Girardin studies a mitochondrial Nod-like receptor (NLR) called NLRX1. While NLRX1 has been implicated in diverse cellular processes, its underlying function remained elusive to researchers until now.
Generally, research in this area revolved around depolarization—the loss of electric potential across the inner mitochondrial membrane—as the major signal for mitochondrial removal.
Girardin and Killackey’s study also showed that depolarization is an upstream cause of restricted mitochondrial protein import.
The findings, published in Molecular Cell, open new avenues for research into diseases where mitochondrial stability is lost.
“This tells us the problem occurs when the protein import fails and the cell receives a signal from the faulty import of the mitochondrial protein, NLRX1. This is the cue to destroy the mitochondria, a process known as mitophagy,” Girardin says.
By looking at the process from a different perspective, the team demonstrated that the established science in this area didn’t show the whole picture.
“We took a step back and connected some of the dots in the literature, which helped us identify that disrupted protein import was a common denominator across many mitochondrial stressors that trigger mitophagy,” says Killackey, a Vanier Scholar who conducted the research during his Ph.D. studies in Girardin’s lab.
The discovery paves the way for researchers to further investigate the role of mitochondrial dysfunction in disease and in metabolically active organs like the brain, heart and kidneys.
“We’ve seen a role for NLRX1-driven mitophagy in muscle function measured through endurance capacity, which could have implications for diseases that involve muscle atrophy or functional deficits. Modifying the extent and efficiency of mitochondrial protein import could also offer therapeutic benefit for neurodegenerative disease,” says Killackey.

The findings are the culmination of 15 years of research, marking a milestone for Girardin.
“I love fundamental questions,” he says. “What happens next with the knowledge is a question of physiology, translational medicine or drug development. So, now it’s time to pass the baton to others, or to partner with enthusiastic collaborators.”
More information:
Samuel A. Killackey et al, Mitochondrial protein import stress regulates the LC3 lipidation step of mitophagy through NLRX1 and RRBP1, Molecular Cell (2022). DOI: 10.1016/j.molcel.2022.06.004
Provided by
University of Toronto
Citation:
Mitochondrial turnover: Researchers discover what causes cell ‘batteries’ to run down (2022, August 22)

