A study at the University of Cologne’s CECAD Cluster of Excellence in Aging Research has identified a protein complex that is activated by defects in the spliceosome, the molecular scissors that process genetic information. Future research could lead to new therapeutic approaches to treat diseases caused by faulty splicing.
The genetic material, in the form of DNA, contains the information that is crucial for the correct functioning of every human and animal cell. From this information repository, RNA, an intermediate between DNA and protein, the functional unit of the cell, is generated. During this process, the genetic information must be tailored for specific cell functions. Information that is not needed (introns) is cut out of the RNA and the important components for proteins (exons) are preserved.
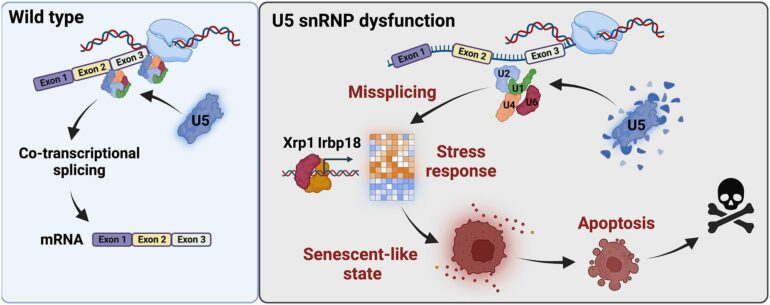
A team of researchers led by Professor Dr. Mirka Uhlirova at the University of Cologne’s CECAD Cluster of Excellence in Aging Research has now discovered that if the processing of this information no longer works properly, a protein complex (C/EBP heterodimer) is activated and directs the cell towards a dormant state, known as cellular senescence. The results appear under the title “Xrp1 governs the stress response program to spliceosome dysfunction” in Nucleic Acids Research.
All eukaryotes (i.e. organisms in which DNA is enclosed within the cell nucleus) have a spliceosome. This is a machine that performs “splicing,” the removal of introns and linking exons to form messenger RNA (mRNA). Malfunctions in the spliceosome lead to diseases known as spliceosomopathies, which may affect many different tissues, and manifest as retinal degeneration or myelodysplastic syndrome, a group of bone marrow diseases affecting the blood.
In the study, the Uhlirova lab used the model organism Drosophila melanogaster, a fruit fly, to investigate how cells within a developing organism respond to spliceosome malfunction. The scientists used a combination of genomics and functional genetics to determine the role of individual genes and interactions among them.
The study showed that cells suffering from a defective spliceosomal U5 snRNP (U5 small nuclear ribonucleoprotein particle) activate a stress signaling response and cellular behaviors that are characteristic of cellular senescence. The senescence program changes crucial functions of the cells. It prevents cells from dividing while stimulating their secretion. Senescence is triggered to preserve cells that are damaged, as their immediate elimination would cause more harm than good. However, senescent cell accumulation can have a negative impact on a tissue as well as the whole organism. Therefore, these cells are ultimately eliminated.
Uhlirova’s team identified the C/EBP-heterodimer protein complex, Xrp1/Irbp18, as the critical driver of the stress response program caused by faulty splicing. Upregulation of Xrp1/Irbp18 in damaged cells led to increased protein production and induced a senescence-like state.
“Senescence is a double-edged sword,” said Uhlirova. One advantage of senescent cells is that they are not all eliminated by cell death at the same time, thus maintaining the integrity of the tissue. After all, partially intact tissue is better than none at all. However, these cells create problems in the long term, as their accumulation promotes disease and aging.
“A functioning spliceosome is a basic prerequisite for healthy cells, tissue and the entire organism,” she concluded. “Additional investigation of the stress signaling program we have identified will be important to further unpack the complex responses triggered by defects in the essential machines controlling gene expression—and how we can influence them.”
In the future, the results could contribute to the development of therapeutic approaches to treat diseases that are caused by malfunctions of the spliceosome.
More information:
Dimitrije Stanković et al, Xrp1 governs the stress response program to spliceosome dysfunction, Nucleic Acids Research (2024). DOI: 10.1093/nar/gkae055
Provided by
University of Cologne
Citation:
Team discovers mechanism that protects tissue after faulty gene expression (2024, February 2)

