
More than 250 million people worldwide have tested positive for SARS-CoV-2, usually after a diagnostic nose swab. Those swabs aren’t trash once they’ve delivered their positive result, though. For scientists like us they carry additional valuable information about the coronavirus. Leftover material from swabs can help us uncover hidden aspects of the COVID-19 pandemic.
Using what are called phylodynamic methods that can track a pathogen’s travels via changes in its genes, researchers are able to pinpoint factors like where and when outbreaks start, the number of undetected infections and common routes of transmission. Phylodynamics can also aid in understanding and tracking the spread of new pathogen variants, such as the recently detected omicron variant of SARS-CoV-2.
What’s in a swab?
Pathogens, just like people, each have a genome. This is RNA or DNA that contains an organism’s genetic code – its instructions for life and the information necessary for reproduction.
It’s now relatively fast and cheap to sequence a pathogen’s genome. In Switzerland, a consortium of government and academic scientists that we’re a part of as already extracted viral genome sequences from almost 80,000 SARS-CoV-2 positive swab tests.
By lining up genetic sequences obtained from different patients, scientists can see which positions in the sequence differ. These differences represent mutations, small errors incorporated into the genome when the pathogen copies itself. We can use these mutational differences as clues to reconstruct chains of transmission and learn about epidemic dynamics along the way.
Phylodynamics: Piecing together genetic clues
Phylodynamic methods provide a way to describe how mutational differences relate to epidemic dynamics. These approaches allow researchers to get from the raw data about where mutations have occurred in the viral or bacterial genome to understanding all the implications. It might sound complicated, but it’s actually pretty easy to give an intuitive idea of how it works.
Mutations in the pathogen genome get passed from person to person in a transmission chain. Many pathogens acquire lots of mutations over the course of an epidemic. Scientists can summarize these mutational similarities and differences using what’s essentially a family tree for the pathogen. Biologists call it a phylogenetic tree. Each branching point represents a transmission event, when the pathogen moved from one person to another.

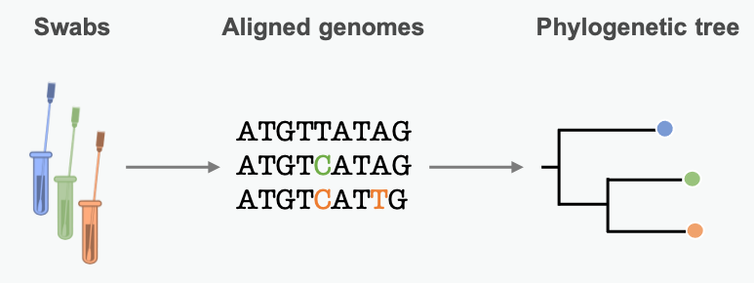
A phylogenetic tree is an approximation of the past transmission chain, based on variations in the pathogen’s genetic sequence.
Guinat, Windels, Nadeau, CC BY-ND
The branch lengths are proportional to the number of differences between sequenced samples. Short branches mean little time between branching points – fast transmission from person to person. Studying the length of branches on this tree can tell us about pathogen spread in the past –…

