An international team of scientists from the University of Luxembourg, Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google has now successfully developed a machine learning algorithm to tackle large and complex quantum systems. The article has been published in Science Advances.
The quantum properties of atoms shape countless biochemical and physical processes. Some of the world’s greatest scientific challenges are fundamentally tied to understanding many interacting atoms over time. These interactions are governed by the laws of quantum mechanics. Examples range from the formation of nucleic acids in the genome, to the decomposition of harmful molecules in the atmosphere.
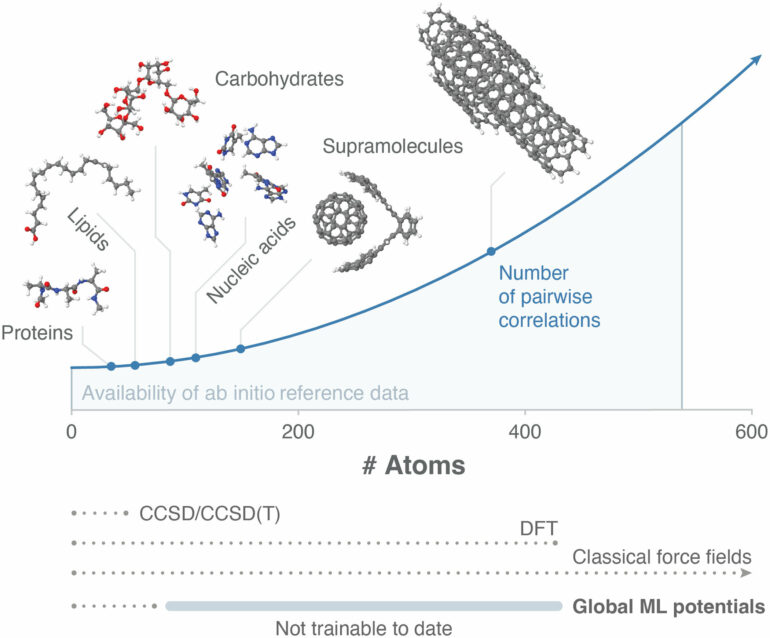
Especially challenging for scientists are the correlations in space and time of such quantum systems: Their most interesting properties do not result from a simple summation of individual contributions from atoms but from intricate atomic correlations. As a result, quantum systems cannot be easily modeled mathematically. In particular, larger quantum systems have so far eluded accurate machine learning (ML) because they cannot be uniquely partitioned into independent small computational packages.
A direct modeling of the complicated correlations would be beyond existing computational capacities.
Realistic and precise
The developed learning algorithm reconstructs so-called global force fields based on machine learning methods without making any potentially undue simplifications. The term “global force fields” describes the approach of considering all atomic Interactions (such as electrostatic, chemical, etc.) of a molecule. It is otherwise common practice to reduce the number of modeled atomic interactions in favor of computational efficiency.
“Quantum states are inseparable and individual constituents cannot act independently without affecting the system as a whole,” explains Prof. Alexandre Tkatchenko, Professor for Theoretical Chemical Physics at the University of Luxembourg. This property marks one of the most sweeping differences between quantum mechanics and the classical Newtonian and electrostatic interactions everyone is intuitively familiar with.
It also poses a dilemma when modeling quantum systems: A ubiquitous paradigm in algorithmic design and an important building block in modeling atomic interactions is to divide a problem into smaller independent parts that are easier to handle for the computer. This is not possible when considering quantum systems due to the properties mentioned above.
Global force fields capable of capturing collective interactions of many atoms in molecular systems currently only scale up to a few dozen atoms using machine learning methods, as model complexity increases significantly with the size of the system at hand. The team addressed this very challenge by developing an algorithm to train global force fields for systems of up to several hundred atoms without ignoring complex correlations.
Their approach carefully separates the strongly coupled atomic interactions within the model into a so-called collective low-dimensional part, which contains recurring interaction patterns, and a so-called residual, which describes the contributions of individual interactions. This separation allows both constituents of the force field reconstruction problem to be solved independently.
The numerical properties of each subproblem, which arise due to unavoidable rounding errors in computer calculations, are specifically taken into consideration. As a consequence, global force fields can be reconstructed based on larger reference data sets to represent more complex interactions, as occur in systems with many atoms or in particularly flexible molecules.
“The numerical characteristics of machine learning algorithms often have a stronger impact than the mathematical formulation suggests, thereby potentially distorting the results. Improvements in numerical stability can have a far-reaching impact on the application of algorithms,” says Dr. Stefan Chmiela, research group leader of the Machine Learning for Many-body Systems group in BIFOLD.
The fact that the developed method can be parallelized across multiple computers is a secondary benefit. It removes algorithmic bottlenecks and enables the effective use of modern parallel computing hardware such as GPUs. “The success of machine learning algorithms is often determined by how efficiently they can run and scale on available hardware,” explains Prof. Dr. Klaus-Robert Müller, Co-Director of BIFOLD.

“This work is a stepping stone to unlock truly predictive quantum simulations of systems with hundreds of atoms,” says Oliver Unke, research scientist at Google. The scientists already successfully performed dynamics simulations of supramolecular complexes on challenging long timescales. Similar simulations are routinely performed in the pharmaceutical industry to identify compounds with specific properties as potential new drug candidates.
“Machine learning methods promise a convergence between exact quantum-mechanical models and efficient empirical solutions. They have the potential to accelerate scientific research in quantum chemistry by offering entirely new opportunities to better understand atomic interactions in intricate physical systems,” explains Alexandre Tkatchenko.
More information:
Stefan Chmiela et al, Accurate global machine learning force fields for molecules with hundreds of atoms, Science Advances (2023). DOI: 10.1126/sciadv.adf0873
Provided by
University of Luxembourg
Citation:
New algorithm enables simulation of complex quantum systems (2023, January 30)

